O albinismo (Albinism) é um grupo de doenças hereditárias causadas por mutações em genes envolvidos na biossíntese ou transporte do pigmento melanina, resultando em deficiência ou ausência de pigmento na pele, cabelo e olhos. Além da deficiência de pigmento, esta doença é caracterizada por anormalidades oculares e das vias ópticas, como hipoplasia foveal, nistagmo e decussação anormal das vias ópticas.
Não há tratamento curativo, e o comprometimento da função visual é não progressivo (estático). No Japão, foi designada como doença rara específica (de acordo com a Lei de Doenças Raras).
O albinismo é classificado em três grupos principais.
Albinismo Oculocutâneo (OCA)
Herança autossômica recessiva: Grupo mais comum. Ocorre deficiência de pigmento na pele, cabelo e olhos.
8 genes e 8 subtipos: OCA1 (TYR), OCA2 (gene OCA2/P), OCA3 (TYRP1), OCA4 (SLC45A2), OCA8 (DCT), entre outros.
Prevalência: Cerca de 1/17.000 no mundo. Em caucasianos, OCA1 representa cerca de 50%; em africanos, OCA2 é o mais comum (1/10.000)1). Em chineses Han, OCA1 é 70,1% e OCA2 é 10,2%1).
Albinismo Ocular (OA)
Herança recessiva ligada ao X: Acomete homens. A pigmentação da pele e cabelo é normal ou levemente reduzida, com achados oculares predominantes.
Mutação no gene GPR143: Envolvido na transdução de sinal da biossíntese de melanossomos. 192 mutações foram relatadas3).
Mulheres portadoras: Apresentam padrão de hipopigmentação em mosaico tipo respingo de lama no fundo de olho. Cerca de 80% apresentam anormalidade do pigmento retiniano3).
Albinismo Sindrômico
Síndrome de Hermansky-Pudlak (HPS): OCA + disfunção plaquetária (tendência a sangramento) + distúrbio da biossíntese de LRO. Existem 11 subtipos4, 5). Pode estar associada a fibrose pulmonar e doença inflamatória intestinal.
Síndrome de Chediak-Higashi (CHS): OCA + imunodeficiência + distúrbio neurológico. Mutação no gene LYST.
12 genes: 12 genes foram identificados para albinismo sindrômico2).
A prevalência mundial é de cerca de 1/17.0001), e na Europa é relatada como cerca de 1/12.0002). Em japoneses, a distribuição conhecida é OCA1 34%, OCA4 27%, HPS1 10%.
QO albinismo é hereditário? Pode ocorrer mesmo que ninguém na família tenha albinismo?
A
OCA é uma doença autossômica recessiva, onde se ambos os pais são portadores de um alelo mutante cada, a probabilidade de a criança ser afetada é de 25%. Pode ocorrer mesmo que os pais sejam fenotipicamente normais, portanto não é incomum mesmo sem histórico familiar. OA é recessiva ligada ao X, afetando principalmente homens, com a mãe sendo portadora. Testes genéticos e aconselhamento genético são importantes.
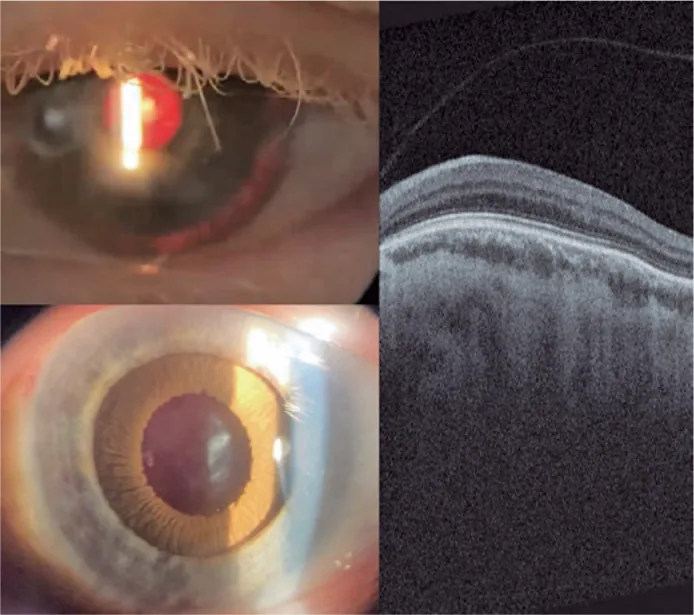
Guilherme Vieira Peixoto; Gabriela Tomaz Martinho; Caio Cezar Toledo de Conti; Eduardo Villaça Filho; Renato Klingelfus Pinheiro. Cataract surgery and artificial iris implantation in patient with oculocutaneous albinism: a case report. Arq Bras Oftalmol.; 87(4):e2022-0286 Figure 4. PMCID: PMC11620172. License: CC BY.
Fotografia com lâmpada de fenda do olho esquerdo 30 dias após a cirurgia, mostrando íris protética no sulco ciliar através de retroiluminação e após midríase farmacológica. OCT de retina mostrando ausência de fosseta foveal e persistência das camadas internas da retina na área esperada da fóvea (hipoplasia macular).
Hipoplasia de fóvea: Fator mais importante para má visão. A ausência de depressão foveal é confirmada por OCT. Na angiografia fluoresceínica, a área avascular perifoveal frequentemente está ausente. No OCA8, todos os casos apresentam grau 3, e o grau se correlaciona com a acuidade visual2).
Transiluminação da íris: Devido à falta de pigmento da íris, observa-se aumento da transiluminação ao exame com lâmpada de fenda. No OCA8, todos os casos são grau 3 2).
Nistagmo (INS): O nistagmo periódico alternante assimétrico é encontrado em cerca de 10% de todos os INS, e atinge até 37% no INS do albinismo 2).
Desvio do trajeto óptico (chiasmal misrouting): As fibras retinianas temporais, que deveriam seguir o trajeto ipsilateral (não cruzado), cruzam excessivamente para o lado contralateral no quiasma óptico. Detectado por VEP. No OCA8, foi relatado coeficiente quiasmático de -0,97/-0,90, indicando assimetria grave 2).
Estrabismo: Ocorre em 53-90,5% dos casos 2).
Fundo de olho hipopigmentado: Devido à falta de pigmento do epitélio pigmentar da retina, os vasos coroidais são visíveis.
Fundo de olho de portadoras de OA1: Em 94% há alterações pigmentares em placas anteriores aos vasos arciformes, e em 74% há transiluminação da íris 3). Manchas de despigmentação na retina periférica (fundo em mosaico) podem ser observadas, sendo úteis para identificação de portadoras.
QA acuidade visual no albinismo muda ao longo da vida?
A
A deficiência visual causada pelo albinismo é estática (não progressiva) e não piora com a idade. No entanto, se a correção refrativa adequada ou o tratamento da ambliopia não forem realizados, a acuidade visual pode permanecer baixa sem melhora. A intervenção precoce com correção refrativa e cuidados de baixa visão é importante.
Abaixo estão os principais genes causadores e características de cada subtipo.
Subtipo
Gene causador
Características principais
OCA1
TYR
Deficiência de tirosinase
OCA2
OCA2 (gene P)
Regulação do pH do melanossomo1)
OCA3
TYRP1
Síntese de eumelanina
OCA4
SLC45A2
Transportador de melanossomo
OCA8
DCT (TYRP2)
Conversão de dopacromo 2)
OA1
GPR143
Sinalização do melanossomo 3)
HPS1/HPS4
HPS1/HPS4
Complexo BLOC-3 4)
HPS11
BLOC1S5
Complexo BLOC-1 5)
Função da proteína OCA2: Pertence à família de antiportadores Na⁺/H⁺ com estrutura de 12 hélices α transmembrana 1). Como componente do canal de ânions específico do melanossomo, regula o pH dos melanossomos estágio I/II através do controle da corrente de cloreto 1). Existem 477 mutações patogênicas registradas no ClinVar 1).
DCT (TYRP2): Catalisa a conversão de dopacromo em DHICA (ácido 5,6-di-hidroxi-indol-2-carboxílico) 2). É o gene causador do OCA8.
GPR143 (OA1): Molécula de sinalização que controla o transporte vesicular do melanossomo. Mutações levam à formação de macromelanossomos 3).
HPS (Complexo BLOC): HPS1/HPS4 funcionam como complexo BLOC-3 como GEF (fator de troca de nucleotídeos guanina) para Rab32/38 4). BLOC-1 consiste em 8 subunidades e está envolvido na reciclagem endossomal, e a mutação do gene BLOC1S5 (BLOS3) causa HPS-11 (descrito pela primeira vez em 2020) 5).
Casamento consanguíneo: A prevalência aumenta em populações com alta taxa de casamentos consanguíneos, como no Paquistão 4).
Diferenças raciais e geográficas: Frequência elevada em algumas regiões da África.
Mutações heterozigóticas compostas (compound heterozygous): Novas mutações heterozigóticas compostas foram identificadas no OCA2, contribuindo para a diversidade fenotípica 1).
Tipo tri-alélico (OCA1): Combinações de três alelos do gene TYR podem causar OCA leve 2).
QPor que é importante saber o tipo de albinismo?
A
Como o prognóstico visual, a presença de complicações sistêmicas e o padrão de herança diferem conforme o tipo, o diagnóstico preciso do subtipo é importante. Especialmente a HPS está associada a tendência a sangramento, fibrose pulmonar e doença inflamatória intestinal, exigindo cuidado antes de cirurgia ou extração dentária. A identificação do subtipo por teste genético está diretamente ligada ao planejamento de manejo adequado.
OCA típico pode ser diagnosticado clinicamente pela combinação de cabelos brancos, pele branca e achados oculares (transiluminação da íris, nistagmo). No entanto, no OA1 japonês, o pigmento permanece na íris, portanto o diagnóstico pode ser atrasado se o paciente consultar apenas com sintomas oculares.
VEP (Potencial Evocado Visual): Mais importante para detectar o cruzamento anormal da via visual. Usar VEP de 3 derivações para avaliar assimetria de ondas entre os hemisférios 2). Quantificar pelo coeficiente quiasmático (-1 assimetria máxima, +1 normal) 2).
Medição do ângulo lambda: Ângulo lambda > 5 graus é um forte indicador clínico de albinismo 2).
Microscopia eletrônica de plaquetas: Usada para diagnóstico definitivo de HPS. Ausência de grânulos densos é achado característico 4).
Teste genético: Sequenciamento do exoma completo (WES) 1) ou sequenciamento de painel 2, 4, 5). Particularmente importante quando o fenótipo é leve ou atípico.
Cirurgia de estrabismo: Realizada para fins estéticos. A cirurgia de Dawson-Trick-Litzkow (DTL) pode ser realizada para redução do nistagmo.
Tratamento do nistagmo: Não há tratamento curativo. A cirurgia de estrabismo é considerada para lidar com a posição anormal da cabeça (garantir a zona nula).
Manejo Cutâneo e Sistêmico
Proteção solar: Protetor solar com bloqueio UVB, roupas e chapéus para proteção da pele. O risco de carcinoma espinocelular deve ser observado mesmo em japoneses.
Manejo da HPS: AINEs (incluindo aspirina) são contraindicados em princípio, pois pioram a disfunção plaquetária. A monitorização regular da função pulmonar a partir da adolescência é recomendada 4).
Aconselhamento genético: Essencial para planejamento familiar, diagnóstico de portadores e determinação do subtipo.
Terapia Medicamentosa para Fibrose Pulmonar na HPS
Para fibrose pulmonar associada à HPS, são utilizados medicamentos antifibróticos.
Liu et al. (2025) relataram um caso de HPS com nova mutação homozigótica em HPS4, no qual a fibrose pulmonar estabilizou por 18 meses após administração de nintedanibe 4). A pirfenidona também é uma opção terapêutica utilizada de forma semelhante.
QA terapia gênica para albinismo está disponível na prática?
A
Atualmente, a terapia gênica para albinismo não está disponível na prática. Terapias gênicas foram aprovadas para outras doenças hereditárias da retina, como retinosquise e LCA (exemplo: Luxturna), e pesquisas para aplicação no albinismo estão em andamento 6). Atualmente, a correção refrativa e os cuidados com baixa visão são o centro do tratamento padrão.
A síntese de melanina segue a seguinte via dentro dos melanossomos:
Tirosina → L-DOPA → DOPA quinona → (eumelanina ou feomelanina)
A conversão de dopacromo em DHICA (ácido 5,6-di-hidroxi-indol-2-carboxílico) é catalisada pela DCT (TYRP2) 2), uma reação essencial para a síntese de eumelanina.
Distúrbio de maturação do melanossomo na OCA2: A redução da função da proteína OCA2 prejudica a transição dos melanossomos para o estágio IV (melanossomos maduros), aumentando os melanossomos imaturos dos estágios I/II 1). O mecanismo principal é a desregulação do pH pelo controle do fluxo de cloreto 1).
Quando a pigmentação do epitélio pigmentar da retina é inadequada, o efeito de bloqueio da luz durante o desenvolvimento é perdido, prejudicando o desenvolvimento morfológico da fóvea (formação da zona avascular e migração densa de cones). Esta é a causa raiz da baixa visão no albinismo.
No albinismo, as fibras da retina temporal cruzam excessivamente para o lado contralateral no quiasma óptico, resultando em mais fibras processadas como fibras nasais. Esse “desvio” pode ser detectado como assimetria do coeficiente quiasmático no VEP2).
No estudo OCA8 de Rateaux et al. (2025), foi utilizado um modelo de camundongo com mutação DCT que reduz a L-DOPA para 50% do tipo selvagem, e a suplementação com L-DOPA reverteu as anormalidades oftalmológicas 2). Isso sugere o papel da L-DOPA como intermediário da síntese de melanina na formação da via óptica.
Mutação GPR143 (OA1): A perda de função da proteína GPR143 prejudica o transporte vesicular dos melanossomas, levando à formação de macromelanossomas 3).
HPS (Distúrbio do complexo BLOC): A perda de função do BLOC-3 prejudica a ativação de Rab32/38, causando falha generalizada na biossíntese de organelas relacionadas aos melanossomas (LRO) 4). Os grânulos densos nas plaquetas e os corpúsculos lamelares nas células epiteliais pulmonares tipo II são afetados. Acredita-se que o acúmulo de substâncias semelhantes a ceroide a partir de corpúsculos lamelares anormais esteja envolvido na fibrose pulmonar 4).
BLOC-1 (HPS-11): Funciona como um complexo de 8 subunidades, incluindo BLOC1S5 (BLOS3), regulando a reciclagem endossomal 5).
7. Pesquisas Recentes e Perspectivas Futuras (Relatos em Fase de Pesquisa)
A L-DOPA é um intermediário da síntese de melanina e desempenha um papel importante no desenvolvimento da retina e na formação da via óptica.
No modelo de camundongo OCA8 de Rateaux et al. (2025), a redução de L-DOPA devido à mutação DCT foi o principal fator do fenótipo oftalmológico, e a suplementação com L-DOPA melhorou as anormalidades 2). A aplicação em humanos está sendo estudada 6).
A avaliação da melhora da função visual com administração de levodopa em pacientes com albinismo está em andamento, com expectativa de efeito promotor do desenvolvimento retiniano 6).
A nitisinona, um inibidor da 4-hidroxifenilpiruvato dioxigenase (4-HPPD), está sendo estudada por sua capacidade de aumentar a melanina intraocular ao modular a via do metabolismo da tirosina 6).
A pesquisa básica sobre terapia de reposição gênica direcionada aos genes relacionados à OCA (TYR, OCA2, etc.) está em andamento. A aprovação do Luxturna (reposição do gene RPE65) para LCA (amaurose congênita de Leber) está acelerando a pesquisa de terapia gênica para doenças hereditárias da retina em geral 6).
Jiang et al. (2024) identificaram uma nova mutação heterozigótica composta (c.635A>G/c.2359+1G>T) no gene OCA2 por sequenciamento completo do exoma (WES) em uma família chinesa 1). A análise funcional confirmou que a proteína mutante apresenta translocação prejudicada para os melanócitos em comparação com o tipo selvagem.
Boeckelmann et al. (2021) detalharam o espectro clínico da HPS-11 devido à mutação BLOC1S5 por meio de um estudo de 5 casos, incluindo aqueles relatados desde 2020 5). Diretrizes de monitoramento dos sintomas pulmonares, oculares e neurológicos foram fornecidas.
A eficácia da terapia antifibrótica com nintedanibe e pirfenidona para fibrose pulmonar na HPS continua sendo avaliada 4).
Flynn et al. (2025) relataram que, no exame de fundo de olho de mulheres portadoras da mutação GPR143 com OA1, 94% apresentaram alterações pigmentares em placas pré-arciformes e 74% apresentaram transiluminação da íris 3). Isso demonstra que anormalidades oculares são frequentes mesmo em portadoras.
QPacientes com albinismo têm alto risco de câncer de pele?
A
Na OCA, devido à falta de pigmento cutâneo, o dano ao DNA causado pela radiação UV se acumula facilmente, aumentando significativamente o risco de câncer de pele, como carcinoma de células escamosas. Acredita-se que o risco seja particularmente alto em pacientes com OCA de ascendência africana. Recomendam-se consultas regulares ao dermatologista e proteção solar rigorosa (protetor solar, roupas de proteção).
Jiang B, Zhang H, Kan Y, Gao X, Du Z, Liu Q. Novel compound heterozygous mutations in OCA2 gene were identified in a Chinese family with oculocutaneous albinism. Mol Genet Genomic Med. 2024;12:e2297. doi:10.1002/mgg3.2297. PMID:37882226; PMCID:PMC10767448.
Rateaux M, Hadj-Rabia S, Barrois R, Zambrowski O, Michaud V, Moreno-Artero E, et al. Chiasmal Decussation in Oculo-Cutaneous Albinism Type 8. Investigative ophthalmology & visual science. 2025;66(2):44. doi:10.1167/iovs.66.2.44. PMID:39951296; PMCID:PMC11824501.
Flynn E, Cheela I, Kaden TR. Female Carrier of Ocular Albinism Linked to Gpr143 Gene. Journal of vitreoretinal diseases. 2025. doi:10.1177/24741264251386389. PMID:41180145; PMCID:PMC12578608.
Liu Q, Qing W, Guo S, Wang Y, Chen Y, Liu J. A novel homozygous HPS4 mutation in Hermansky-Pudlak syndrome: case report and literature review. Therapeutic advances in respiratory disease. 2025;19:17534666251383677. doi:10.1177/17534666251383677. PMID:41277635; PMCID:PMC12644420.
Boeckelmann D, Wolter M, Kasmann-Kellner B, Koehler U, Schieber-Nakamura L, Zieger B. A novel likely pathogenic variant in the BLOC1S5 gene associated with Hermansky-Pudlak syndrome type 11 and an overview of human BLOC-1 deficiencies. Cells. 2021;10:2630. doi:10.3390/cells10102630. PMID:34685610; PMCID:PMC8533863.
Gurnani B, et al. Nystagmus review. Clin Ophthalmol. 2025;19:1617-1648.
Copie o texto do artigo e cole no assistente de IA de sua preferência.
Artigo copiado para a área de transferência
Abra um assistente de IA abaixo e cole o texto copiado na conversa.