Альбинизм — это группа наследственных заболеваний, вызванных мутациями генов, участвующих в биосинтезе или транспорте меланина, что приводит к отсутствию или уменьшению пигментации кожи, волос и глаз. Помимо дефицита пигмента, для этой группы заболеваний характерны специфические аномалии глаз и зрительных путей, такие как гипоплазия фовеа, нистагм и аномальный перекрест зрительных путей.
Радикального лечения не существует, нарушения зрительной функции являются непрогрессирующими (стационарными). В Японии это заболевание отнесено к категории редких заболеваний (Закон о редких заболеваниях).
Аутосомно-рецессивное наследование: самая частая группа. Дефицит пигмента в коже, волосах и глазах.
8 генов, 8 подтипов: ГКА1 (TYR), ГКА2 (ген OCA2/P), ГКА3 (TYRP1), ГКА4 (SLC45A2), ГКА8 (DCT) и др.
Распространенность: около 1/17 000 в мире. У европеоидов ГКА1 составляет около 50%, у африканцев наиболее част ГКА2 (1/10 000)1). У китайцев хань ГКА1 составляет 70,1%, ГКА2 – 10,2%1).
Глазной альбинизм (ГА)
X-сцепленное рецессивное наследование: встречается у мужчин. Пигментация кожи и волос нормальная или слегка снижена, преобладают глазные проявления.
Мутация гена GPR143: участвует в передаче сигналов биосинтеза меланосом. Сообщается о 192 мутациях3).
Женщины-носители: на глазном дне наблюдается мозаичный рисунок гипопигментации, напоминающий брызги грязи. Примерно у 80% выявляются аномалии пигментации сетчатки3).
Синдромальный альбинизм
Синдром Хермански-Пудлака (HPS): ГКА + дисфункция тромбоцитов (склонность к кровотечениям) + нарушение биосинтеза LRO. Существует 11 подтипов4, 5). Может осложняться легочным фиброзом и воспалительными заболеваниями кишечника.
12 генов: для синдромального альбинизма идентифицировано 12 генов2).
Мировая распространенность составляет около 1/17 0001), в Европе сообщается о примерно 1/12 0002). У японцев известно распределение: ГКА1 – 34%, ГКА4 – 27%, HPS1 – 10%.
QПередается ли альбинизм по наследству? Может ли он возникнуть, если в семье нет больных альбинизмом?
A
OCA наследуется по аутосомно-рецессивному типу: если оба родителя являются носителями одного мутантного аллеля, вероятность развития заболевания у ребенка составляет 25%. Поскольку родители могут быть фенотипически нормальными, отсутствие семейного анамнеза не редкость. OA наследуется по X-сцепленному рецессивному типу, в основном поражает мужчин, мать является носителем. Генетическое тестирование и генетическое консультирование важны.
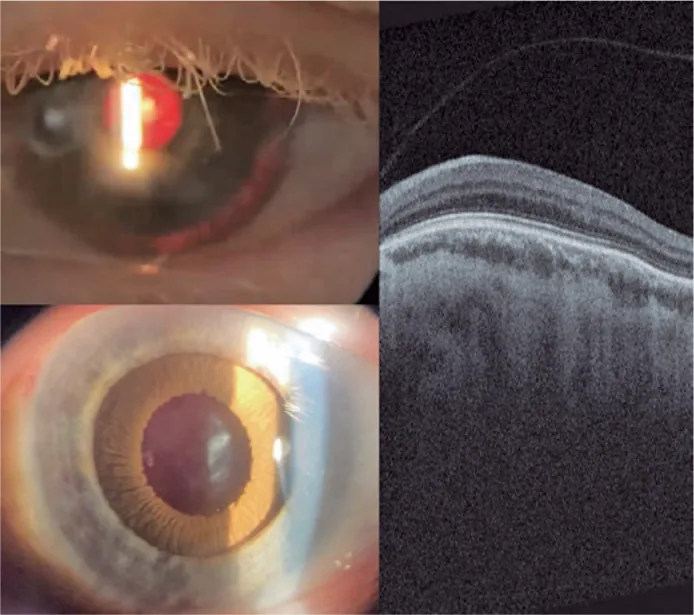
Guilherme Vieira Peixoto; Gabriela Tomaz Martinho; Caio Cezar Toledo de Conti; Eduardo Villaça Filho; Renato Klingelfus Pinheiro. Cataract surgery and artificial iris implantation in patient with oculocutaneous albinism: a case report. Arq Bras Oftalmol.; 87(4):e2022-0286 Figure 4. PMCID: PMC11620172. License: CC BY.
Фотография левого глаза с помощью щелевой лампы через 30 дней после операции, показывающая протез радужки на цилиарной борозде при ретроиллюминации и после медикаментозного мидриаза. ОКТ сетчатки показывает отсутствие фовеальной ямки и сохранение внутренних слоев сетчатки в области ожидаемой фовеи (макулярная гипоплазия).
Гипоплазия фовеа: важнейший фактор плохого зрения. ОКТ подтверждает отсутствие фовеального углубления. При флуоресцентной ангиографии часто отсутствует бессосудистая зона вокруг макулы. При ОСА8 все случаи имеют 3-ю степень, и степень коррелирует с остротой зрения2).
Трансиллюминация радужки: из-за недостатка пигмента радужки при биомикроскопии наблюдается усиление трансиллюминации. При ОСА8 все случаи имеют 3-ю степень2).
Нистагм (INS): асимметричный периодический альтернирующий нистагм встречается примерно в 10% всех INS, а при альбинизме INS — до 37%2).
Аномальное перекрещивание зрительных путей (chiasmal misrouting): височные волокна сетчатки, которые в норме следуют ипсилатеральным (неперекрещенным) путем, чрезмерно перекрещиваются на противоположную сторону в хиазме. Выявляется с помощью ВПЗП. При ОСА8 сообщается о коэффициенте хиазмы −0,97/−0,90, что указывает на выраженную асимметрию2).
Косоглазие: наблюдается в 53–90,5% случаев2).
Гипопигментированное глазное дно: из-за недостатка пигмента в пигментном эпителии сетчатки просвечивают сосуды хориоидеи.
Глазное дно носителей ОА1: у 94% наблюдаются полосовидные пигментные изменения кпереди от аркад, у 74% — трансиллюминация радужки3). На периферии сетчатки могут быть депигментированные пятна (мозаичное глазное дно), что полезно для идентификации носителей.
QМеняется ли острота зрения при альбинизме в течение жизни?
A
Нарушения зрения, вызванные альбинизмом, являются стационарными (непрогрессирующими) и не ухудшаются с возрастом. Однако, если не проводить соответствующую коррекцию рефракции или лечение амблиопии, острота зрения может оставаться фиксированной без улучшения. Важно раннее вмешательство с коррекцией рефракции и помощью при слабом зрении.
Основные гены-причины и характеристики каждого подтипа приведены ниже.
Подтип
Ген-причина
Основные характеристики
OCA1
TYR
Дефицит тирозиназы
OCA2
OCA2 (P-ген)
Регуляция pH меланосом1)
OCA3
TYRP1
Синтез эумеланина
OCA4
SLC45A2
Транспортер меланосом
OCA8
DCT (TYRP2)
Превращение допахрома2)
OA1
GPR143
Сигнал меланосом3)
HPS1/HPS4
HPS1/HPS4
Комплекс BLOC-34)
HPS11
BLOC1S5
Комплекс BLOC-15)
Функция белка OCA2 : Он принадлежит к семейству антипортеров Na⁺/H⁺ с 12-трансмембранной α-спиральной структурой 1). Как компонент меланосом-специфичного анионного канала, он регулирует pH меланосом стадии I/II посредством контроля хлоридного тока 1). ClinVar содержит 477 патогенных мутаций 1).
DCT (TYRP2) : Катализирует превращение допахрома в DHICA (5,6-дигидроксииндол-2-карбоновую кислоту) 2). Это ген-причина OCA8.
GPR143 (OA1) : Сигнальная молекула, регулирующая везикулярный транспорт меланосом. Мутации приводят к образованию макромеланосом 3).
HPS (комплекс BLOC) : HPS1/HPS4 функционируют как комплекс BLOC-3, действующий как GEF (гуанин-нуклеотидный обменный фактор) для Rab32/38 4). BLOC-1 состоит из 8 субъединиц и участвует в эндосомальном рециклинге; мутация гена BLOC1S5 (BLOS3) вызывает HPS-11 (впервые описан в 2020 году) 5).
Кровнородственные браки : В популяциях с высокой частотой кровнородственных браков, например в Пакистане, распространенность повышается 4).
Этнические и региональные различия : В некоторых регионах Африки частота высока.
Компаунд-гетерозиготные мутации : При OCA2 выявлены новые компаунд-гетерозиготные мутации, способствующие фенотипическому разнообразию 1).
Триаллельный тип (OCA1) : Комбинации трех аллелей гена TYR могут приводить к легкой форме OCA 2).
QПочему важно знать тип альбинизма?
A
Прогноз зрения, наличие системных осложнений и тип наследования различаются в зависимости от типа, поэтому важна точная диагностика подтипа. В частности, синдром Хермански-Пудлака (HPS) сопровождается склонностью к кровотечениям, фиброзом легких и воспалительным заболеванием кишечника, что требует осторожности перед операцией или удалением зуба. Идентификация подтипа с помощью генетического тестирования напрямую связана с разработкой соответствующего плана ведения.
Типичный ОКА можно диагностировать клинически по сочетанию белых волос, белой кожи и глазных признаков (просвечивание радужки, нистагм). Однако при японском ОА1 пигмент сохраняется в радужке, поэтому при обращении только с глазными симптомами диагноз может быть поставлен с задержкой.
Щелевая лампа : Подтверждение просвечивания радужки. Оценка степени.
ОКТ (оптическая когерентная томография) : Обязательна для градации фовеальной гипоплазии. Выполнима даже у детей, полезна для принятия решения о раннем вмешательстве6).
ВП (зрительные вызванные потенциалы) : Наиболее важны для выявления аномального перекреста зрительных путей. Оценка асимметрии волн между левым и правым полушариями с помощью 3-канального ВП2). Количественная оценка с помощью коэффициента перекреста ( −1 максимальная асимметрия, +1 норма)2).
Измерение угла лямбда : Угол лямбда > 5 градусов является сильным клиническим признаком альбинизма2).
Электронная микроскопия тромбоцитов : Используется для окончательной диагностики HPS. Характерно отсутствие плотных гранул4).
Генетическое тестирование : Секвенирование всего экзома (WES)1) или панельное секвенирование2, 4, 5). Особенно важно при легком или атипичном фенотипе.
Радикального лечения не существует. Цель лечения — максимальное улучшение зрительной функции и контроль осложнений.
Ведение зрения
Коррекция рефракции : Ношение очков с раннего детства является основой. Раннее вмешательство для предотвращения амблиопии важно.
Светозащитные очки : Используйте линзы, блокирующие около 20% света в помещении и около 80% на улице 5).
Контактные линзы с радужкой : Могут использоваться для косметического улучшения и уменьшения светобоязни.
Помощь при слабовидении : Используйте вспомогательные средства, такие как лупы, увеличивающие устройства для чтения и планшеты.
Нистагм и косоглазие
Операция при косоглазии : Проводится с косметической целью. Иногда выполняется операция Доусона-Трика-Литцкова (DTL) для уменьшения нистагма.
Лечение нистагма : Этиотропного лечения нет. Хирургия косоглазия может рассматриваться для коррекции аномального положения головы (нулевой зоны).
Уход за кожей и системное лечение
Защита от солнца : Защита кожи с помощью солнцезащитного крема, блокирующего УФБ, одежды и головных уборов. Даже у японцев следует обращать внимание на риск плоскоклеточного рака.
Ведение HPS : НПВП (включая аспирин) обычно противопоказаны, так как ухудшают функцию тромбоцитов. Рекомендуется регулярный мониторинг функции легких с подросткового возраста 4).
Генетическое консультирование : Необходимо для планирования семьи, диагностики носительства и определения подтипа.
При легочном фиброзе, ассоциированном с HPS, используются антифибротические препараты.
Liu и соавт. (2025) сообщили о случае HPS с новой гомозиготной мутацией HPS4, при котором введение нинтеданиба стабилизировало легочный фиброз в течение 18 месяцев 4). Пирфенидон также является используемым вариантом лечения.
QДоступна ли генная терапия альбинизма в клинической практике?
A
На данный момент генная терапия альбинизма еще не внедрена в клиническую практику. Для других наследственных заболеваний сетчатки, таких как ретиношизис и LCA, одобрены методы генной терапии (например, Luxturna), и ведутся исследования по их применению при альбинизме6). В настоящее время основой стандартного лечения являются коррекция рефракции и помощь при слабом зрении.
Синтез меланина в меланосомах происходит по следующему пути:
Тирозин → L-ДОФА → ДОФА-хинон → (эумеланин или феомеланин)
Превращение допахрома в DHICA (5,6-дигидроксииндол-2-карбоновую кислоту) катализируется DCT (TYRP2)2), что является обязательной реакцией для синтеза эумеланина.
Нарушение созревания меланосом при OCA2 : Снижение функции белка OCA2 приводит к нарушению перехода меланосом в стадию IV (зрелые меланосомы) и увеличению количества незрелых меланосом стадий I/II1). Основным механизмом является нарушение регуляции pH за счет контроля хлоридного тока1).
При недостаточной пигментации пигментного эпителия сетчатки теряется светозащитный эффект в процессе развития, что препятствует морфологическому развитию фовеа (формированию бессосудистой зоны и плотной миграции колбочек). Это является основной причиной плохого зрения при альбинизме.
При альбинизме волокна из височной части сетчатки чрезмерно перекрещиваются на противоположную сторону в хиазме, так что большее количество волокон обрабатывается как носовые. Этот «misrouting» регистрируется на ВВП как асимметрия хиазмального коэффициента2).
В исследовании OCA8 Rateaux и соавт. (2025) использовалась мышиная модель с мутацией DCT, снижающей уровень L-DOPA до 50% от дикого типа, и было показано, что добавление L-DOPA устраняет офтальмологические аномалии 2). Это указывает на роль L-DOPA как промежуточного продукта синтеза меланина в формировании зрительного пути.
Мутация GPR143 (OA1) : Потеря функции белка GPR143 нарушает везикулярный транспорт меланосом, что приводит к образованию макромеланосом 3).
HPS (нарушения комплекса BLOC) : Потеря функции BLOC-3 нарушает активацию Rab32/38, что приводит к общему нарушению биогенеза органелл, связанных с меланосомами (LRO: лизосомоподобные органеллы) 4). Поражаются плотные тельца тромбоцитов и ламеллярные тельца в эпителиальных клетках легких II типа. Считается, что в основе легочного фиброза лежит накопление цероидоподобных веществ из аномальных ламеллярных телец 4).
BLOC-1 (HPS-11) : Функционирует как комплекс из 8 субъединиц, включая BLOC1S5 (BLOS3), и регулирует эндосомальный рециклинг 5).
7. Новейшие исследования и перспективы (исследовательская стадия)
L-DOPA является промежуточным продуктом синтеза меланина и играет важную роль в развитии сетчатки и формировании зрительного пути.
На мышиной модели OCA8 Rateaux и соавт. (2025) снижение L-DOPA из-за мутации DCT было основным фактором офтальмологического фенотипа, и добавление L-DOPA улучшило аномалии 2). Изучается применение у человека 6).
Проводится оценка улучшения зрительной функции при введении леводопы пациентам с альбинизмом, ожидается стимулирующий эффект на развитие сетчатки 6).
Нитизинон, ингибитор 4-гидроксифенилпируватдиоксигеназы (4-HPPD), исследуется на предмет возможности увеличения внутриглазного меланина путем регуляции пути метаболизма тирозина 6).
Фундаментальные исследования генной заместительной терапии, нацеленной на гены, ассоциированные с ОКА (TYR, OCA2 и др.), продолжаются. Одобрение Luxturna (замещение гена RPE65) для LCA (врожденный амавроз Лебера) ускоряет исследования генной терапии наследственных заболеваний сетчатки в целом 6).
Jiang и соавт. (2024) с помощью полного экзомного секвенирования (WES) идентифицировали новую компаунд-гетерозиготную мутацию (c.635A>G/c.2359+1G>T) гена OCA2 в китайской семье 1). Функциональный анализ подтвердил, что мутантный белок имеет нарушение транслокации в меланоциты по сравнению с диким типом.
Boeckelmann и соавт. (2021) подробно описали клинический спектр HPS-11, вызванного мутацией BLOC1S5, в исследовании 5 случаев, включая случаи, зарегистрированные после 2020 года 5). Представлены рекомендации по мониторингу легочных, глазных и неврологических симптомов.
Эффективность антифибротической терапии нинтеданибом и пирфенидоном при легочном фиброзе HPS продолжает оцениваться 4).
Flynn и соавт. (2025) сообщили, что при исследовании глазного дна у женщин-носителей OA1 с мутацией GPR143 у 94% были выявлены дугообразные макулярные пигментные изменения, а у 74% - транслюминесценция радужки 3). Было показано, что офтальмологические аномалии часто встречаются даже у носителей.
QПовышен ли риск рака кожи у пациентов с альбинизмом?
A
При ОКА из-за отсутствия кожного пигмента легче накапливаются повреждения ДНК, вызванные ультрафиолетом, что значительно повышает риск рака кожи, такого как плоскоклеточный рак. Считается, что риск особенно высок у пациентов с ОКА африканского происхождения. Рекомендуются регулярные осмотры дерматолога и тщательная защита от солнца (солнцезащитный крем, закрытая одежда).
Jiang B, Zhang H, Kan Y, Gao X, Du Z, Liu Q. Novel compound heterozygous mutations in OCA2 gene were identified in a Chinese family with oculocutaneous albinism. Mol Genet Genomic Med. 2024;12:e2297. doi:10.1002/mgg3.2297. PMID:37882226; PMCID:PMC10767448.
Rateaux M, Hadj-Rabia S, Barrois R, Zambrowski O, Michaud V, Moreno-Artero E, et al. Chiasmal Decussation in Oculo-Cutaneous Albinism Type 8. Investigative ophthalmology & visual science. 2025;66(2):44. doi:10.1167/iovs.66.2.44. PMID:39951296; PMCID:PMC11824501.
Flynn E, Cheela I, Kaden TR. Female Carrier of Ocular Albinism Linked to Gpr143 Gene. Journal of vitreoretinal diseases. 2025. doi:10.1177/24741264251386389. PMID:41180145; PMCID:PMC12578608.
Liu Q, Qing W, Guo S, Wang Y, Chen Y, Liu J. A novel homozygous HPS4 mutation in Hermansky-Pudlak syndrome: case report and literature review. Therapeutic advances in respiratory disease. 2025;19:17534666251383677. doi:10.1177/17534666251383677. PMID:41277635; PMCID:PMC12644420.
Boeckelmann D, Wolter M, Kasmann-Kellner B, Koehler U, Schieber-Nakamura L, Zieger B. A novel likely pathogenic variant in the BLOC1S5 gene associated with Hermansky-Pudlak syndrome type 11 and an overview of human BLOC-1 deficiencies. Cells. 2021;10:2630. doi:10.3390/cells10102630. PMID:34685610; PMCID:PMC8533863.
Gurnani B, et al. Nystagmus review. Clin Ophthalmol. 2025;19:1617-1648.
Скопируйте текст статьи и вставьте его в выбранный ИИ-ассистент.
Статья скопирована в буфер обмена
Откройте ИИ-ассистент ниже и вставьте скопированный текст в чат.