Albinisme adalah kelompok penyakit genetik yang disebabkan oleh mutasi gen yang terlibat dalam biosintesis atau transportasi pigmen melanin, mengakibatkan kekurangan atau tidak adanya pigmen pada kulit, rambut, dan mata. Selain defisiensi pigmen, penyakit ini ditandai dengan kelainan mata dan jalur penglihatan yang khas seperti hipoplasia fovea, nistagmus, dan dekusasi abnormal jalur penglihatan.
Tidak ada pengobatan kuratif, dan gangguan fungsi penglihatan bersifat non-progresif (statis). Di Jepang, penyakit ini telah ditetapkan sebagai penyakit langka tertentu (berdasarkan Undang-Undang Penyakit Langka).
Albinisme diklasifikasikan menjadi tiga kelompok utama.
Albinisme Okulokutaneus (OCA)
Warisan resesif autosomal: Kelompok yang paling umum. Terjadi defisiensi pigmen pada kulit, rambut, dan mata.
8 gen dan 8 subtipe: OCA1 (TYR), OCA2 (gen OCA2/P), OCA3 (TYRP1), OCA4 (SLC45A2), OCA8 (DCT), dan lainnya.
Prevalensi: Sekitar 1/17.000 di dunia. Pada orang Kaukasia, OCA1 mencakup sekitar 50%, sedangkan pada orang Afrika, OCA2 paling umum (1/10.000)1). Pada etnis Han Tiongkok, OCA1 70,1% dan OCA2 10,2%1).
Albinisme Okular (OA)
Warisan resesif terkait-X: Terjadi pada pria. Pigmen kulit dan rambut normal atau sedikit berkurang, dengan temuan okular dominan.
Mutasi gen GPR143: Terlibat dalam transduksi sinyal biosintesis melanosom. 192 mutasi telah dilaporkan3).
Wanita pembawa: Menunjukkan pola hipopigmentasi mosaik seperti percikan lumpur di fundus. Sekitar 80% memiliki kelainan pigmen retina3).
Albinisme Sindromik
Sindrom Hermansky-Pudlak (HPS): OCA + disfungsi trombosit (kecenderungan perdarahan) + gangguan biosintesis LRO. Terdapat 11 subtipe4, 5). Dapat disertai fibrosis paru dan penyakit radang usus.
Sindrom Chediak-Higashi (CHS): OCA + defisiensi imun + gangguan neurologis. Mutasi gen LYST.
12 gen: 12 gen telah diidentifikasi untuk albinisme sindromik2).
Prevalensi dunia sekitar 1/17.0001), dan di Eropa dilaporkan sekitar 1/12.0002). Pada orang Jepang, distribusi yang diketahui adalah OCA1 34%, OCA4 27%, HPS1 10%.
QApakah albinisme bersifat herediter? Dapatkah terjadi meskipun tidak ada anggota keluarga yang menderita albinisme?
A
OCA adalah kelainan resesif autosomal, di mana jika kedua orang tua masing-masing membawa satu alel mutan (karier), maka kemungkinan anak terkena adalah 25%. Karena dapat terjadi meskipun kedua orang tua normal secara fenotip, hal ini tidak jarang terjadi tanpa riwayat keluarga. OA adalah resesif terkait-X yang terutama menyerang laki-laki, dengan ibu sebagai karier. Tes genetik dan konseling genetik penting.
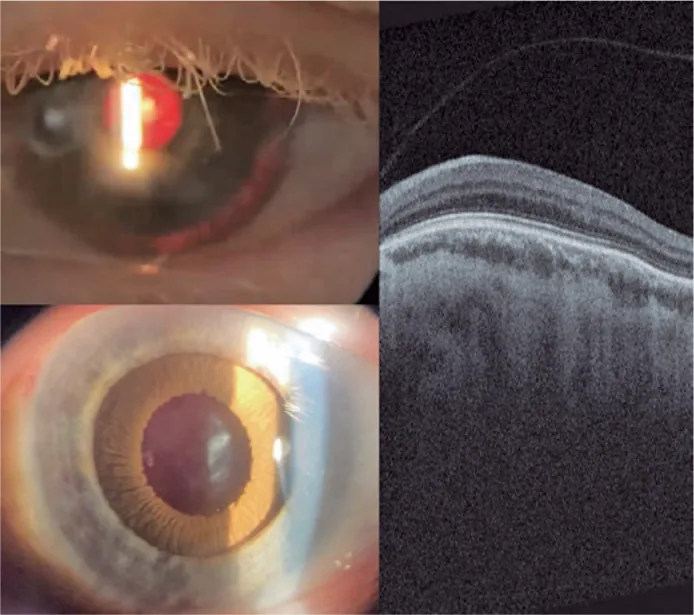
Guilherme Vieira Peixoto; Gabriela Tomaz Martinho; Caio Cezar Toledo de Conti; Eduardo Villaça Filho; Renato Klingelfus Pinheiro. Cataract surgery and artificial iris implantation in patient with oculocutaneous albinism: a case report. Arq Bras Oftalmol.; 87(4):e2022-0286 Figure 4. PMCID: PMC11620172. License: CC BY.
Foto slit-lamp mata kiri 30 hari setelah operasi, menunjukkan iris prostetik pada sulkus siliaris melalui retroiluminasi dan setelah midriasis farmakologis. OCTretina menunjukkan tidak adanya foveal pit dan persistensi lapisan dalam retina di area yang diharapkan sebagai fovea (hipoplasia makula).
Hipoplasia fovea: Faktor terpenting untuk penglihatan buruk. Tidak adanya cekungan fovea dikonfirmasi dengan OCT. Pada angiografi fluorescein, area avaskular perifoveal sering tidak terlihat. Pada OCA8, semua kasus menunjukkan grade 3, dan grade berkorelasi dengan ketajaman penglihatan2).
Transiluminasi iris: Karena kurangnya pigmen iris, peningkatan transiluminasi terlihat pada pemeriksaan slit-lamp. Pada OCA8, semua kasus grade 3 2).
Nistagmus (INS): Nistagmus periodik bergantian asimetris ditemukan pada sekitar 10% dari seluruh INS, dan mencapai 37% pada INS albinisme 2).
Penyimpangan jalur optik (chiasmal misrouting): Serat retina temporal, yang seharusnya mengikuti jalur ipsilateral (tidak menyilang), menyilang secara berlebihan ke sisi kontralateral di kiasma optikum. Dideteksi dengan VEP. Pada OCA8, dilaporkan koefisien kiasma -0.97/-0.90, menunjukkan asimetri berat 2).
Fundus pembawa OA1: Pada 94% ditemukan perubahan pigmen bercak di anterior pembuluh arkuata, dan pada 74% ditemukan transiluminasi iris3). Bercak depigmentasi di retina perifer (fundus mosaik) kadang ditemukan, berguna untuk identifikasi pembawa.
QApakah ketajaman penglihatan pada albinisme berubah sepanjang hidup?
A
Gangguan penglihatan akibat albinisme bersifat statis (non-progresif) dan tidak memburuk seiring bertambahnya usia. Namun, jika koreksi refraksi yang tepat atau terapi ambliopia tidak dilakukan, ketajaman penglihatan mungkin tetap buruk tanpa perbaikan. Intervensi dini dengan koreksi refraksi dan perawatan low vision sangat penting.
Berikut adalah gen penyebab utama dan karakteristik setiap subtipe.
Subtipe
Gen penyebab
Karakteristik utama
OCA1
TYR
Defisiensi tirosinase
OCA2
OCA2 (gen P)
Regulasi pH melanosom1)
OCA3
TYRP1
Sintesis eumelanin
OCA4
SLC45A2
Transporter melanosom
OCA8
DCT (TYRP2)
Konversi dopakrom 2)
OA1
GPR143
Sinyal melanosom 3)
HPS1/HPS4
HPS1/HPS4
Kompleks BLOC-3 4)
HPS11
BLOC1S5
Kompleks BLOC-1 5)
Fungsi protein OCA2: Termasuk dalam keluarga antiporter Na⁺/H⁺ dengan struktur heliks α transmembran 12 1). Sebagai komponen saluran anion spesifik melanosom, mengatur pH melanosom tahap I/II melalui kontrol arus klorida 1). Terdapat 477 mutasi patogenik terdaftar di ClinVar 1).
DCT (TYRP2): Mengkatalisis konversi dopakrom menjadi DHICA (asam 5,6-dihidroksiindol-2-karboksilat) 2). Merupakan gen penyebab OCA8.
GPR143 (OA1): Molekul sinyal yang mengontrol transpor vesikel melanosom. Mutasi menyebabkan pembentukan makromelanosom 3).
HPS (Kompleks BLOC): HPS1/HPS4 berfungsi sebagai kompleks BLOC-3 sebagai GEF (faktor pertukaran nukleotida guanin) untuk Rab32/38 4). BLOC-1 terdiri dari 8 subunit dan terlibat dalam daur ulang endosom, dan mutasi gen BLOC1S5 (BLOS3) menyebabkan HPS-11 (pertama kali dijelaskan pada 2020) 5).
Perkawinan sedarah: Prevalensi meningkat pada populasi dengan tingkat perkawinan sedarah tinggi seperti Pakistan 4).
Perbedaan ras dan geografis: Frekuensi tinggi di beberapa wilayah Afrika.
Mutasi heterozigot majemuk (compound heterozygous): Mutasi heterozigot majemuk baru telah diidentifikasi pada OCA2, berkontribusi pada keragaman fenotipe 1).
Tipe tri-alel (OCA1): Kombinasi tiga alel gen TYR dapat menyebabkan OCA ringan 2).
QMengapa penting mengetahui tipe albinisme?
A
Karena prognosis penglihatan, adanya komplikasi sistemik, dan pola pewarisan berbeda tergantung tipe, diagnosis subtipe yang akurat sangat penting. Terutama HPS disertai kecenderungan perdarahan, fibrosis paru, dan penyakit radang usus, sehingga diperlukan kehati-hatian sebelum operasi atau pencabutan gigi. Identifikasi subtipe melalui tes genetik langsung terkait dengan penyusunan rencana tata laksana yang tepat.
OCA tipikal dapat didiagnosis secara klinis dari kombinasi rambut putih, kulit putih, dan temuan okular (transiluminasi iris, nistagmus). Namun, pada OA1 Jepang, pigmen masih tersisa di iris, sehingga diagnosis dapat tertunda jika pasien datang hanya dengan gejala okular.
OCT (Optical Coherence Tomography): Penting untuk grading hipoplasia fovea. Dapat dilakukan pada anak-anak, membantu menentukan intervensi dini 6).
VEP (Visual Evoked Potential): Paling penting untuk mendeteksi crossing abnormal jalur visual. Gunakan VEP 3-lead untuk menilai asimetri gelombang antar hemisfer 2). Kuantifikasi dengan koefisien kiasma (-1 asimetri maksimal, +1 normal) 2).
Pengukuran sudut lambda: Sudut lambda > 5 derajat merupakan indikator klinis kuat untuk albinisme 2).
Fluorescein angiography (FA): Memperjelas pola mosaik fundus pada pembawa OA1.
Mikroskop elektron trombosit: Digunakan untuk diagnosis pasti HPS. Tidak adanya dense body merupakan temuan khas 4).
Tes genetik: Whole exome sequencing (WES) 1) atau panel sequencing 2, 4, 5). Sangat penting jika fenotip ringan atau atipikal.
Tidak ada terapi kuratif. Tujuan terapi adalah memaksimalkan fungsi penglihatan dan mengelola komplikasi.
Manajemen Visual
Koreksi refraksi: Kacamata sejak bayi adalah dasar. Intervensi dini untuk mencegah ambliopia penting.
Kacamata pelindung cahaya: Gunakan lensa yang memblokir sekitar 20% cahaya di dalam ruangan dan sekitar 80% di luar ruangan 5).
Lensa kontak berwarna: Dapat digunakan untuk perbaikan kosmetik dan mengurangi fotofobia.
Perawatan low vision: Gunakan alat bantu seperti kaca pembesar, alat baca pembesar, dan tablet.
Nistagmus dan Strabismus
Operasi strabismus: Dilakukan untuk tujuan kosmetik. Operasi Dawson-Trick-Litzkow (DTL) kadang dilakukan untuk mengurangi nistagmus.
Terapi nistagmus: Tidak ada pengobatan kuratif. Operasi strabismus dipertimbangkan untuk mengatasi posisi kepala abnormal (memastikan null zone).
Manajemen Kulit dan Sistemik
Perlindungan matahari: Tabir surya yang memblokir UVB, pakaian, dan topi untuk melindungi kulit. Risiko karsinoma sel skuamosa perlu diwaspadai bahkan pada orang Jepang.
Manajemen HPS: NSAID (termasuk aspirin) pada prinsipnya kontraindikasi karena memperburuk disfungsi trombosit. Pemantauan fungsi paru secara teratur sejak remaja dianjurkan 4).
Konseling genetik: Penting untuk perencanaan keluarga, diagnosis karier, dan penentuan subtipe.
Untuk fibrosis paru yang terkait dengan HPS, digunakan obat antifibrotik.
Liu dkk. (2025) melaporkan kasus HPS dengan mutasi homozigot baru pada HPS4, di mana fibrosis paru stabil selama 18 bulan setelah pemberian nintedanib 4). Pirfenidone juga merupakan pilihan terapi yang digunakan serupa.
QApakah terapi gen untuk albinisme sudah tersedia secara praktis?
A
Saat ini, terapi gen untuk albinisme belum tersedia secara praktis. Terapi gen telah disetujui untuk penyakit retina herediter lainnya seperti retinoskisis dan LCA (contoh: Luxturna), dan penelitian untuk aplikasi pada albinisme sedang berlangsung 6). Saat ini, koreksi refraksi dan perawatan low vision adalah inti dari terapi standar.
Sintesis melanin mengikuti jalur berikut di dalam melanosom:
Tirosin → L-DOPA → DOPA kuinon → (eumelanin atau feomelanin)
Konversi dopakrom menjadi DHICA (asam 5,6-dihidroksiindol-2-karboksilat) dikatalisis oleh DCT (TYRP2) 2), reaksi yang penting untuk sintesis eumelanin.
Gangguan pematangan melanosom pada OCA2: Penurunan fungsi protein OCA2 menghambat transisi melanosom ke tahap IV (melanosom matang), meningkatkan jumlah melanosom imatur tahap I/II 1). Mekanisme utamanya adalah gangguan regulasi pH melalui kontrol arus klorida 1).
Jika pigmentasi epitel pigmen retina tidak memadai, efek penghalang cahaya selama perkembangan hilang, menghambat perkembangan morfologis fovea (pembentukan zona avaskular dan migrasi padat sel kerucut). Ini adalah akar penyebab penglihatan buruk pada albinisme.
Pada albinisme, serat dari retina temporal menyilang secara berlebihan ke sisi kontralateral di kiasma optikum, sehingga lebih banyak serat diproses sebagai serat nasal. “Misrouting” ini dapat dideteksi sebagai asimetri koefisien kiasma pada VEP2).
Dalam studi OCA8 oleh Rateaux dkk. (2025), digunakan model tikus dengan mutasi DCT yang menurunkan L-DOPA hingga 50% dari tipe liar, dan suplementasi L-DOPA terbukti memulihkan kelainan oftalmologis 2). Hal ini menunjukkan peran L-DOPA sebagai prekursor sintesis melanin dalam pembentukan jalur visual.
Mutasi GPR143 (OA1): Kehilangan fungsi protein GPR143 mengganggu transpor vesikel melanosom, menyebabkan pembentukan makromelanosom 3).
HPS (Gangguan kompleks BLOC): Kehilangan fungsi BLOC-3 mengganggu aktivasi Rab32/38, menyebabkan kegagalan biosintesis organel terkait melanosom (LRO) secara umum 4). Badan padat dalam trombosit dan badan lamela dalam sel epitel paru tipe II terpengaruh. Akumulasi zat seperti seroid dari badan lamela abnormal diduga berperan dalam fibrosis paru 4).
BLOC-1 (HPS-11): Berfungsi sebagai kompleks 8 subunit termasuk BLOC1S5 (BLOS3), mengatur daur ulang endosom 5).
7. Penelitian Terkini dan Prospek Masa Depan (Laporan Tahap Penelitian)
L-DOPA adalah prekursor sintesis melanin dan berperan penting dalam perkembangan retina serta pembentukan jalur visual.
Pada model tikus OCA8 oleh Rateaux dkk. (2025), penurunan L-DOPA akibat mutasi DCT merupakan faktor utama fenotip oftalmologis, dan suplementasi L-DOPA memperbaiki kelainan tersebut 2). Aplikasi pada manusia sedang diteliti 6).
Evaluasi perbaikan fungsi visual dengan pemberian levodopa pada pasien albinisme sedang berlangsung, dengan harapan efek stimulasi perkembangan retina6).
Nitisinone, penghambat 4-hidroksifenilpiruvat dioksigenase (4-HPPD), diteliti kemampuannya untuk meningkatkan melanin intraokular dengan memodulasi jalur metabolisme tirosin 6).
Penelitian dasar terapi penggantian gen yang menargetkan gen terkait OCA (TYR, OCA2, dll.) sedang berlangsung. Persetujuan Luxturna (penggantian gen RPE65) untuk LCA (amaurosis kongenital Leber) mempercepat penelitian terapi gen untuk penyakit retina herediter secara umum 6).
Jiang dkk. (2024) mengidentifikasi mutasi heterozigot majemuk baru (c.635A>G/c.2359+1G>T) pada gen OCA2 melalui sekuensing eksom utuh (WES) pada keluarga Tionghoa 1). Analisis fungsional mengonfirmasi bahwa protein mutan mengalami gangguan translokasi ke melanosit dibandingkan dengan tipe liar.
Boeckelmann dkk. (2021) merinci spektrum klinis HPS-11 akibat mutasi BLOC1S5 melalui studi 5 kasus termasuk yang dilaporkan sejak 2020 5). Pedoman pemantauan gejala paru, mata, dan saraf telah diberikan.
Efektivitas terapi antifibrosis dengan nintedanib dan pirfenidon untuk fibrosis paru HPS terus dievaluasi 4).
Flynn dkk. (2025) melaporkan bahwa pada pemeriksaan fundus wanita pembawa mutasi GPR143 dengan OA1, 94% menunjukkan perubahan pigmen bercak pre-arkuat, dan 74% menunjukkan transiluminasi iris3). Hal ini menunjukkan bahwa kelainan okular sering terjadi bahkan pada pembawa.
QApakah pasien albinisme berisiko tinggi terkena kanker kulit?
A
Pada OCA, karena kurangnya pigmen kulit, kerusakan DNA akibat sinar UV mudah terakumulasi, sehingga risiko kanker kulit seperti karsinoma sel skuamosa meningkat secara signifikan. Risiko dianggap lebih tinggi pada pasien OCA keturunan Afrika. Kunjungan rutin ke dokter kulit dan perlindungan matahari yang ketat (tabir surya, pakaian pelindung) dianjurkan.
Jiang B, Zhang H, Kan Y, Gao X, Du Z, Liu Q. Novel compound heterozygous mutations in OCA2 gene were identified in a Chinese family with oculocutaneous albinism. Mol Genet Genomic Med. 2024;12:e2297. doi:10.1002/mgg3.2297. PMID:37882226; PMCID:PMC10767448.
Rateaux M, Hadj-Rabia S, Barrois R, Zambrowski O, Michaud V, Moreno-Artero E, et al. Chiasmal Decussation in Oculo-Cutaneous Albinism Type 8. Investigative ophthalmology & visual science. 2025;66(2):44. doi:10.1167/iovs.66.2.44. PMID:39951296; PMCID:PMC11824501.
Flynn E, Cheela I, Kaden TR. Female Carrier of Ocular Albinism Linked to Gpr143 Gene. Journal of vitreoretinal diseases. 2025. doi:10.1177/24741264251386389. PMID:41180145; PMCID:PMC12578608.
Liu Q, Qing W, Guo S, Wang Y, Chen Y, Liu J. A novel homozygous HPS4 mutation in Hermansky-Pudlak syndrome: case report and literature review. Therapeutic advances in respiratory disease. 2025;19:17534666251383677. doi:10.1177/17534666251383677. PMID:41277635; PMCID:PMC12644420.
Boeckelmann D, Wolter M, Kasmann-Kellner B, Koehler U, Schieber-Nakamura L, Zieger B. A novel likely pathogenic variant in the BLOC1S5 gene associated with Hermansky-Pudlak syndrome type 11 and an overview of human BLOC-1 deficiencies. Cells. 2021;10:2630. doi:10.3390/cells10102630. PMID:34685610; PMCID:PMC8533863.
Gurnani B, et al. Nystagmus review. Clin Ophthalmol. 2025;19:1617-1648.
Salin teks artikel dan tempelkan ke asisten AI pilihan Anda.
Artikel disalin ke papan klip
Buka asisten AI di bawah, lalu tempelkan teks yang disalin ke kotak chat.