A sarcoidose é uma doença inflamatória crônica idiopática caracterizada por granulomas não caseosos. Pulmões, pele e olhos são os órgãos mais afetados, mas quando atinge o sistema nervoso central (SNC) e/ou periférico (SNP) é chamada de neurosarcoidose (NS). Pode acompanhar sarcoidose de outros órgãos ou afetar apenas o sistema nervoso.
Incidência: Nos EUA, 11 por 100.000 em brancos, 35,5-36 por 100.000 em afro-americanos
Prevalência: Cerca de 152-215 por 100.000 pessoas
Idade mais comum: 30-50 anos. Mais comum em mulheres afro-americanas
Frequência de envolvimento neurológico: Ocorre em 5-15% dos pacientes com sarcoidose4)7). Na autópsia, há evidências em até 25%, sugerindo possíveis casos latentes
Início com sintomas neurológicos: Em cerca de 70% dos casos, os sintomas neurológicos são o início, muitas vezes sem diagnóstico sistêmico prévio3)
No Japão, a sarcoidose é relativamente comum e é a principal causa de uveíte. Em homens, é mais frequente na faixa dos 20 anos; em mulheres, há dois picos: aos 20 anos e aos 50-60 anos.
QCom que frequência ocorre a sarcoidose neurológica?
A
Lesões neurológicas são encontradas em 5-15% dos pacientes com sarcoidose4)7). Na autópsia, sinais de sarcoidose neurológica são encontrados em até 25%, indicando muitos casos latentes. Em cerca de 70% dos casos, os sintomas neurológicos são o primeiro sinal da doença3).
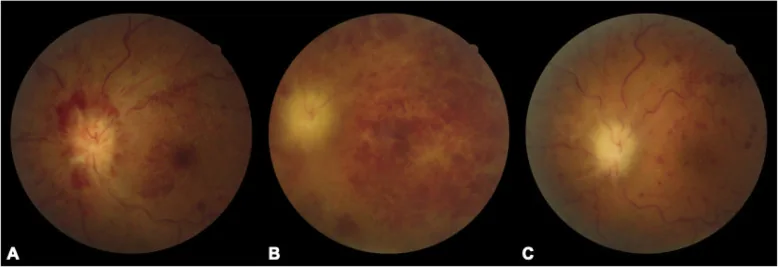
Chaoyi Feng, Qian Chen, Wei Liu et al. Neurosarcoidosis presenting as CRVO combined CRAO: a biopsy-proven case report of a Chinese patient. BMC Ophthalmology. 2020 Aug 27; 20:348. Figure 1. PMCID: PMC7457306. License: CC BY.
Realce leptomeníngeo difuso: Achado mais frequente na RM com contraste gadolínio.
Realce de nervos cranianos: Realce contrastado dos nervos cranianos, incluindo facial e óptico.
Lesões cerebrais pseudotumorais: Únicas ou múltiplas. Facilmente confundidas com tumores ou metástases.
Hidrocefalia: Ambos os tipos comunicante e não comunicante. Presente em 57% dos casos2).
Achados Oculares (6 critérios IWOS)
Uveíte anterior granulomatosa: Precipitados ceráticos gordurosos, nódulos de íris.
Nódulos angulares ou sinéquias anteriores periféricas em tenda: Achados característicos do segmento anterior.
Opacidades vítreas em grumos: Opacidades em forma de bola de neve ou rosário.
Periflebite retiniana: Inflamação perivascular e nódulos ocorrendo principalmente em veias.
Múltiplas manchas exsudativas coriorretinianas semelhantes a cera: ou lesões atróficas semelhantes a manchas de fotocoagulação.
Granuloma do disco óptico ou granuloma coroidal: Vermelhidão e inchaço do disco óptico.
4 padrões de RM da medula espinhal (PMC11213433)6)
Mielite transversa de segmento longo (LETM): 45%. Lesão transversa extensa longitudinal de C2 ao cone medular.
Meningorradiculite: 23%.
Mielite pseudotumoral: 23%.
Mielite anterior: 10%. Ocorre em áreas adjacentes à degeneração discal.
Achados acompanhantes de referência para sarcoidose ocular: Ceratoconjuntivite seca, episclerite/esclerite, aumento da glândula lacrimal, paralisia do nervo facial.
Características da neuropatia óptica: 28% ocorrem bilateralmente de forma sequencial, 37% com edema do disco óptico, 4% com perineurite óptica10).
QQuais sintomas aparecem nos olhos?
A
A uveíte é a mais comum, caracterizada por uveíte anterior granulomatosa com precipitados ceráticos gordurosos e nódulos de íris. Suspeita-se de sarcoidose ocular quando há 2 ou mais dos 6 critérios IWOS (uveíte anterior granulomatosa, nódulos do ângulo, opacidades vítreas em bola, perivasculite retiniana, manchas exsudativas coriorretinianas semelhantes a cera, granuloma do disco óptico/coroidal). Na neuropatia óptica, 28% ocorrem bilateralmente10).
A etiologia da NS é desconhecida. A patologia básica é a formação de granulomas não caseosos devido a uma reação imune celular Th1 (alergia tipo IV). Postula-se uma hiperativação de macrófagos e células T devido à exposição prolongada a estímulos antigênicos. No Japão, há relatos sugerindo o envolvimento de Propionibacterium acnes.
O diagnóstico de NS é feito combinando diversos quadros clínicos e resultados de exames. O diagnóstico definitivo requer confirmação tecidual de granuloma não caseoso, mas devido aos riscos da biópsia do SNC, múltiplos critérios diagnósticos são utilizados.
ACE sérico: Elevação útil para auxílio diagnóstico (ex.: 73 U/L9)). Útil para sarcoidose sistêmica, mas especificidade insuficiente para o tipo neurológico
Lisozima sérica: Elevação observada
sIL-2R sérico: Elevado. Juntamente com linfopenia, é um marcador eficaz para sarcoidose ocular
Cintilografia com 67Ga-citrato ou FDG-PET: Captação positiva auxilia no diagnóstico
Ressonância magnética com gadolínio: Maior sensibilidade. O achado mais comum é realce leptomeníngeo difuso/espessado. Distribuição periventricular + realce leptomeníngeo é a combinação típica de dois achados 1)
Tomografia computadorizada de tórax: Para confirmação de BHL. Em brancos, a sarcoidose não pode ser descartada mesmo com TC de tórax normal
FDG-PET: Útil para avaliar lesões em outros locais e selecionar sítio de biópsia 3)
Ressonância magnética de parede vascular (VWI): Útil para avaliar vasculite associada à neurosarcoidose
A confirmação de granuloma não caseoso é o padrão-ouro. A biópsia do SNC é ideal, mas invasiva; portanto, biópsias de outros locais, como linfonodo, pele, conjuntiva ou biópsia pulmonar transbrônquica, são consideradas como alternativas.
Meningite tuberculosa, esclerose múltipla, linfoma do SNC, granulomatose linfomatoide associada ao EBV
Granulomatose com poliangiite (GPA), meningioma, glioma óptico, doença de Behçet, doença relacionada a IgG4
QQuais exames são necessários para o diagnóstico definitivo de neurosarcoidose?
A
O diagnóstico definitivo requer confirmação tecidual de granuloma não caseoso por biópsia do SNC. No entanto, devido à sua invasividade, é frequentemente utilizado um diagnóstico escalonado que combina exame do líquido cefalorraquidiano (aumento de proteínas, pleocitose linfocítica), RM com contraste de gadolínio, ACE e sIL-2R séricos, e TC de tórax (confirmação de BHL). Se o granuloma não caseoso for confirmado em outros locais (linfonodos, pele, biópsia transbrônquica, etc.), o diagnóstico Provável pode ser estabelecido.
Por ser uma doença com atividade flutuante, em casos leves pode-se esperar melhora espontânea e apenas observar com colírios de corticosteroide. Em casos graves, administra-se corticosteroide sistêmico.
Inflamação do Segmento Anterior
Colírio de corticosteroide: Rinderon 0,1% 4 vezes ao dia. Continuar mesmo sem inflamação da câmara anterior para prevenir nódulos do ângulo.
Midriáticos: Midrin P 3 vezes ao dia (para prevenir sinéquia posterior).
Inflamação do Segmento Posterior (Casos Graves)
Corticosteroide oral: Prednisolona 0,5-1,0 mg/kg/dia, com redução gradual.
Abaixo está um exemplo de prescrição para redução gradual da prednisolona.
Período
Dose
2 semanas
30 mg/dia
1 mês
20 mg/dia
1 mês
15 mg/dia
1 mês
10 mg/dia
1 mês
7,5 mg/dia
1 mês
5 mg/dia
1 mês
5 mg em dias alternados
Injeção sub-Tenon posterior: Esteroide de liberação prolongada (Kenacort A 40 mg). Eficaz para edema macular cistoide e opacidade vítrea. Pico de efeito em cerca de 1 mês, duração do efeito em cerca de 3 meses
Tratamento das complicações oculares
Catarata associada: Cirurgia na fase de remissão. Pode ser realizada sob corticosteroide oral
Glaucoma secundário: Colírios hipotensores (preparações de PG, betabloqueadores, inibidores da anidrase carbônica, agonistas do receptor α2) → inibidores da anidrase carbônica orais → infusão de D-manitol. Cirurgia (trabeculectomia) é especialmente eficaz no glaucoma esteroidal
Áreas avasculares por vasculite oclusiva: Fotocoagulação retiniana
Casos com vasculite cerebral: A combinação de glicocorticoide + MTX/ciclofosfamida/infliximabe é a principal estratégia terapêutica7). A razão de risco de eventos cerebrovasculares em 5 anos após o diagnóstico de sarcoidose é de 10,06, significativamente elevada7).
Epilepsia associada: Usar antiepiléptico como levetiracetam em combinação 5).
No hipopituitarismo total devido a lesão hipofisária, é necessária reposição hormonal com hidrocortisona, levotiroxina e desmopressina (para diabetes insípido). O dano neurológico é frequentemente irreversível e pode exigir reposição ao longo da vida 4).
QQuanto tempo dura o tratamento com esteroides?
A
Varia muito conforme a atividade da doença. Na sarcoidose ocular, tipicamente começa com prednisolona 30 mg e reduz gradualmente para 5 mg em dias alternados ao longo de cerca de 7 meses. Em casos graves ou recorrentes, a dose de manutenção (5-10 mg/dia) pode ser necessária a longo prazo. A sarcoidose neurológica frequentemente alterna recaídas e remissões, sendo essencial o acompanhamento a longo prazo.
As células Th1 liberam IL-2 e IFN-γ, recrutando e ativando macrófagos. Os macrófagos ativados secretam citocinas e mantêm a formação contínua de granulomas. Histologicamente, células epitelioides e células gigantes multinucleadas formam o centro, com linfócitos, plasmócitos e mastócitos se acumulando ao redor. Corpúsculos asteroides podem ser observados dentro das células gigantes multinucleadas.
Granulomas se formam dentro ou ao redor da parede vascular. Ocorrem preferencialmente em pequenas artérias perfurantes. A razão de risco de eventos cerebrovasculares em 5 anos após o diagnóstico de sarcoidose é de 10,06, indicando risco acentuadamente elevado de complicações cerebrovasculares 7).
A destruição inflamatória da mielina é o principal componente. Na biópsia neuromuscular, confirmam-se granulomas epineurais e infiltração endoneural 6).
Ocorre obstrução do fluxo do LCR devido à cicatrização granulomatosa das meninges e comprometimento da absorção do LCR devido à inflamação das vilosidades aracnoides. Ambos os tipos, comunicante e não comunicante, podem ocorrer 2).
A infiltração granulomatosa no hipotálamo, haste hipofisária e hipófise prejudica o eixo endócrino. Em casos graves, pode ocorrer diabetes insípido central com Na 168 (hipernatremia) 4).
7. Pesquisas Recentes e Perspectivas Futuras (Relatos em Fase de Pesquisa)
Espera-se que a técnica de imagem da parede vascular por RM melhore a precisão diagnóstica da vasculite associada à neurosarcoidose.
Na revisão sistemática de Focke et al. (2025), a detecção de lesões na parede vascular por RM VWI foi bem-sucedida em 9 de 13 pacientes com NS (69%) 7). Pode ser capaz de visualizar inflamação da parede vascular que é difícil de capturar pela RM convencional com contraste de gadolínio.
Como biomarcadores de NS associada a vasculite, a neopterina e a lisozima no LCR estão chamando a atenção.
Focke et al. (2025) relataram que a neopterina no LCR estava elevada em 100% dos casos (média 5,2 ng/ml) e a lisozima em 75% (média 4,25 mg/l) em casos de NS associada a vasculite 7). Esses marcadores podem se tornar ferramentas diagnósticas.
Como imagem independente de FDG-PET, a imagem de receptor de somatostatina e a imagem PET direcionada a CXCR4 (expresso em macrófagos ativados) estão sendo exploradas.
Ach T, Ben Yahia W, Halloul I, Sghaier F, Atig A. Neurosarcoidosis-Induced Hypophysitis Mimicking Pituitary Macroadenoma. Cureus. 2023;15(6):e39865. doi:10.7759/cureus.39865. PMID:37404438; PMCID:PMC10315063.
Kafai Golahmadi A, Craven CL, Watkins LD. Neurosarcoidosis Mimicking Normal Pressure Hydrocephalus. Cureus. 2023;15(6):e40281. doi:10.7759/cureus.40281. PMID:37448383; PMCID:PMC10336621.
Hanif Z, Gonzalez Ramos KN, Razminia P, Aigbe E, Ghafourian P. A Perplexing Case of Bladder Mass Biopsy-Proven Neurosarcoidosis. Cureus. 2023;15(6):e40865. doi:10.7759/cureus.40865. PMID:37489187; PMCID:PMC10363404.
Khawaja MA, Awesat BE, Yasini MN, et al. Neurosarcoidosis Presented as an Isolated Brain Lesion. Cureus. 2023;15(9):e45837. doi:10.7759/cureus.45837.
Chaubey M, Meena K, Singh T, Reddy S, Raj R, Chaudhary A, et al. Neurosarcoidosis: An under-diagnosed cause of myelopathy. Journal of family medicine and primary care. 2024;13(5):2157-2160. doi:10.4103/jfmpc.jfmpc_987_23. PMID:38948561; PMCID:PMC11213433.
Focke JK, Brokbals M, Becker J, Veltkamp R, van de Beek D, Brouwer MC, et al. Cerebral vasculitis related to neurosarcoidosis: a case series and systematic literature review. Journal of neurology. 2025;272(2):135. doi:10.1007/s00415-024-12868-2. PMID:39812656; PMCID:PMC11735521.
Ryan Shields, Olivia Sagan, Logan Roebke, Josh Vander Maten, Shailen Shah, George Chang, Dalia Ibrahim, Sumayya Naz. Rare case of multifocal extradural and intramedullary neurosarcoidosis without pulmonary involvement: a case report and literature review. Spinal Cord Ser Cases. 2021;7(1). doi:10.1038/s41394-021-00450-1.
Sarac E, Erzurum SA, Arif A. An Unusual Presentation of Neurosarcoidosis. Am J Case Rep. 2022;23:e937125. doi:10.12659/ajcr.937125.
Desmond PK, Ben JB, Elizabeth MG, Gordon TP. Optic neuropathy associated with systemic sarcoidosis. Neurol Neuroimmunol Neuroinflammation. 2016;3:e270.
Kidd D, Beynon HL.. The neurological complications of systemic sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis. 2003;20(2):85-94. PMID:12870717.
Copie o texto do artigo e cole no assistente de IA de sua preferência.
Artigo copiado para a área de transferência
Abra um assistente de IA abaixo e cole o texto copiado na conversa.