Die Sarkoidose ist eine chronische entzündliche Erkrankung unbekannter Ursache, die durch nicht-verkäsende Granulome gekennzeichnet ist. Lunge, Haut und Augen sind die häufigsten Organe, aber die Beteiligung des zentralen Nervensystems (ZNS) und/oder des peripheren Nervensystems (PNS) wird als Neurosarkoidose (NS) bezeichnet. Sie kann mit einer systemischen Sarkoidose einhergehen oder isoliert das Nervensystem betreffen.
Inzidenz : In den USA 11 pro 100.000 bei Weißen, 35,5–36 pro 100.000 bei Afroamerikanern
Prävalenz : etwa 152–215 pro 100.000 Personen
Bevorzugtes Alter : 30–50 Jahre. Häufiger bei afroamerikanischen Frauen
Häufigkeit neurologischer Läsionen : Bei 5–15 % der Sarkoidose-Patienten festgestellt4)7). Bei Autopsien finden sich in bis zu 25 % Hinweise, möglicherweise latent
Erstmanifestation mit neurologischen Symptomen : In etwa 70 % der Fälle treten neurologische Symptome zuerst auf, oft ohne vorherige systemische Diagnose3)
In Japan ist Sarkoidose relativ häufig und die häufigste Ursache für Uveitis. Männer sind häufiger in den 20ern betroffen, Frauen zeigen zwei Gipfel in den 20ern und zwischen 50 und 60 Jahren.
QWie häufig tritt Neurosarkoidose auf?
A
Bei 5–15 % der Sarkoidose-Patienten werden neurologische Läsionen festgestellt4)7). Bei Autopsien finden sich in bis zu 25 % Anzeichen einer Neurosarkoidose, was auf eine hohe Anzahl latenter Fälle hindeutet. In etwa 70 % der Fälle sind neurologische Symptome das erste Anzeichen der Erkrankung3).
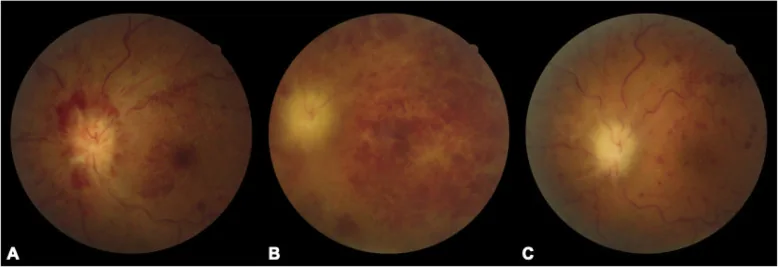
Chaoyi Feng, Qian Chen, Wei Liu et al. Neurosarcoidosis presenting as CRVO combined CRAO: a biopsy-proven case report of a Chinese patient. BMC Ophthalmology. 2020 Aug 27; 20:348. Figure 1. PMCID: PMC7457306. License: CC BY.
Kammerwinkelknötchen oder zeltförmige periphere anteriore Synechien : charakteristische Vorderabschnittsbefunde.
Klumpige Glaskörpertrübungen : schneeball- oder perlenkettenartige Trübungen.
Retinale Perivaskulitis : Perivaskulitis und Knötchen hauptsächlich an Venen.
Multiple wachsartige chorioidale exsudative Flecken : oder atrophische Herde ähnlich wie Photokoagulationsnarben.
Optikusneuritis-Granulom oder chorioidales Granulom : Rötung und Schwellung der Papille.
4 Muster der spinalen MRT (PMC11213433)6)
Longitudinally extensive transverse myelitis (LETM) : 45 %. Längs ausgedehnte transversale Läsion von C2 bis zum Conus medullaris.
Meningomyelitis : 23 %.
Tumorähnliche Myelitis : 23 %.
Vordere Myelitis : 10 %. Tritt in angrenzenden Bereichen der Bandscheibendegeneration auf.
Begleitbefunde, die auf eine okuläre Sarkoidose hinweisen : Keratokonjunktivitis sicca, Episkleritis/Skleritis, Tränendrüsenschwellung, Fazialisparese.
Merkmale der Optikusneuropathie : 28 % treten bilateral aufeinanderfolgend auf, 37 % zeigen eine Papillenschwellung, 4 % eine Optikusperineuritis10).
QWelche Symptome treten am Auge auf?
A
Uveitis ist am häufigsten, gekennzeichnet durch eine granulomatöse anteriore Uveitis mit Präzipitaten wie Hammelfett und Irisknötchen. Zwei oder mehr der 6 IWOS-Kriterien (granulomatöse anteriore Uveitis, Kammerwinkelknötchen, klumpige Glaskörpertrübungen, retinale Perivaskulitis, wachsartige chorioidale exsudative Flecken, Papillen-/Chorioidalgranulom) lassen eine okuläre Sarkoidose vermuten. Bei Optikusneuropathie treten 28 % bilateral auf10).
Die Ätiologie der Sarkoidose ist ungeklärt. Die Bildung nicht-verkäsender Granulome durch eine Th1-Zell-vermittelte Immunantwort (Typ-IV-Allergie) ist die grundlegende Pathologie. Eine Überaktivierung von Makrophagen und T-Zellen durch langfristige Exposition gegenüber antigenen Reizen wird angenommen. In Japan gibt es Berichte, die eine Beteiligung von Propionibacterium acnes zeigen.
Die Diagnose der NS wird durch die Kombination verschiedener klinischer Bilder und Untersuchungsergebnisse gestellt. Für die definitive Diagnose ist der histologische Nachweis nicht-verkäsender Granulome erforderlich, aber da eine ZNS-Biopsie risikobehaftet ist, werden mehrere Diagnosekriterien verwendet.
Serum-ACE : Erhöhung ist diagnostisch hilfreich (z. B. 73 U/L9)). Nützlich für systemische Sarkoidose, aber Spezifität für die neurologische Form unzureichend
Serum-Lysozym : Erhöhung feststellbar
Serum-löslicher IL-2R (sIL-2R) : Erhöhung. Zusammen mit Lymphopenie ein wirksamer Marker für okuläre Sarkoidose
67Ga-Citrat-Szintigraphie oder FDG-PET : positive Anreicherung unterstützt die Diagnose
Der Nachweis eines nicht-verkäsenden Granuloms ist der Goldstandard. Eine ZNS-Biopsie ist ideal, aber invasiv; Alternativen wie Lymphknoten-, Haut-, Konjunktival- oder transbronchiale Lungenbiopsie werden in Betracht gezogen.
QWelche Untersuchungen sind für die definitive Diagnose einer Neurosarkoidose erforderlich?
A
Die definitive Diagnose erfordert den histologischen Nachweis nicht-verkäsender Granulome durch eine ZNS-Biopsie. Aufgrund der Invasivität wird jedoch häufig ein schrittweiser diagnostischer Ansatz verwendet, der Liquoranalyse (Protein erhöht, Lymphozytose), Gadolinium-verstärktes MRT, Serum-ACE und sIL-2R sowie CT-Thorax (Bestätigung von BHL) kombiniert. Wenn nicht-verkäsende Granulome an anderen Stellen (Lymphknoten, Haut, transbronchiale Biopsie usw.) bestätigt werden, ist eine wahrscheinliche Diagnose möglich.
Die Krankheit verläuft in Schüben. Bei leichten Fällen kann eine spontane Besserung abgewartet werden, und eine Überwachung nur mit steroidhaltigen Augentropfen ist möglich. Schwere Fälle werden systemisch mit Kortikosteroiden behandelt.
Entzündung des vorderen Augenabschnitts
Steroid-Augentropfen : Rinderon 0,1% 4-mal täglich. Auch ohne Vorderkammerentzündung zur Vorbeugung von Kammerwinkelknötchen fortsetzen.
Mydriatika : Mydrin P 3-mal täglich (Vorbeugung von hinteren Synechien).
Entzündung des hinteren Augenabschnitts (schwere Fälle)
Orale Steroide : Prednisolon 0,5–1,0 mg/kg/Tag als Anfangsdosis, dann ausschleichend.
Ein Beispiel für ein Prednisolon-Ausschleichschema ist unten dargestellt.
Zeitraum
Dosis
2 Wochen
30 mg/Tag
1 Monat
20 mg/Tag
1 Monat
15 mg/Tag
1 Monat
10 mg/Tag
1 Monat
7,5 mg/Tag
1 Monat
5 mg/Tag
1 Monat
5 mg jeden zweiten Tag
Hintere Tenon-Kapsel-Injektion: Depot-Steroid (Kenacort A 40 mg). Wirksam bei zystoidem Makulaödem und Glaskörpertrübung. Wirkungsgipfel nach etwa 1 Monat, Wirkungsdauer etwa 3 Monate.
Behandlung von Augenkomplikationen
Begleitkatarakt : Operation in der Entzündungsphase. Kann unter oralen Steroiden durchgeführt werden.
Sekundärglaukom : Drucksenkende Augentropfen (PG-Analoga, Betablocker, Carboanhydrasehemmer, Alpha-2-Agonisten) → orale Carboanhydrasehemmer → D-Mannitol-Infusion. Operation (Trabekulektomie) besonders wirksam bei Steroidglaukom.
Avaskuläre Areale durch obliterative Vaskulitis : Netzhautphotokoagulation.
Infliximab : TNF-α-Inhibitor. Auch bei begleitender zerebraler Vaskulitis einsetzbar.
Adalimumab : Ebenfalls ein TNF-α-Inhibitor. Zunehmender Einsatz bei therapierefraktären Fällen.
Fälle mit zerebraler Vaskulitis : Kombination aus Glukokortikoid + MTX/Cyclophosphamid/Infliximab als Hauptbehandlungsstrategie7). Das Hazard-Ratio für zerebrovaskuläre Ereignisse innerhalb von 5 Jahren nach Sarkoidose-Diagnose beträgt 10,06 und ist signifikant erhöht7).
Epilepsie-Komorbidität: Antiepileptikum wie Levetiracetam zusätzlich verabreichen5).
Hydrozephalus: VP-Shunt (einstellbares Ventil 5 cmH2O + Antisiphonvorrichtung) in Kombination mit Steroiden ist wirksam2). Tritt postoperativ ein vorübergehender rascher Ventrikelkollaps (Slit-Ventrikel) auf, wird das Ventil auf 15 cmH2O umgestellt2).
Rückenmarkskompression (extradurale Läsion): Chirurgische Dekompression (Laminektomie) kann erforderlich sein8).
Sofern keine lebensbedrohliche Komplikation vorliegt, sind Operationsindikationen grundsätzlich begrenzt.
Bei Panhypopituitarismus aufgrund einer Hypophysenläsion ist eine Hormonsubstitution mit Hydrocortison, Levothyroxin und Desmopressin (Diabetes insipidus) erforderlich. Nervenschäden sind oft irreversibel und können eine lebenslange Substitution erforderlich machen4).
QWie lange dauert die Steroidbehandlung?
A
Das hängt stark von der Krankheitsaktivität ab. Bei okulärer Sarkoidose wird Prednisolon typischerweise mit 30 mg begonnen und über etwa 7 Monate auf 5 mg jeden zweiten Tag reduziert. Bei schweren oder rezidivierenden Fällen kann eine Erhaltungsdosis (5–10 mg/Tag) langfristig erforderlich sein. Die Neurosarkoidose verläuft oft schubförmig mit Remissionen, sodass eine langfristige Nachbeobachtung unerlässlich ist.
Th1-Zellen setzen IL-2 und IFN-γ frei, rekrutieren und aktivieren Makrophagen. Aktivierte Makrophagen sezernieren Zytokine und erhalten die anhaltende Granulombildung aufrecht. Histologisch bilden Epitheloidzellen und mehrkernige Riesenzellen das Zentrum, umgeben von Lymphozyten, Plasmazellen und Mastzellen. In den mehrkernigen Riesenzellen können gelegentlich Asteroidkörper beobachtet werden.
Granulome bilden sich innerhalb oder um die Gefäßwand, besonders in kleinen perforierenden Arterien. Das Hazard-Ratio für zerebrovaskuläre Ereignisse innerhalb von 5 Jahren nach der Sarkoidose-Diagnose beträgt 10,06, was auf ein deutlich erhöhtes Risiko für zerebrovaskuläre Komplikationen hinweist 7).
Es kommt zu einer Obstruktion des Liquorflusses durch granulomatöse Vernarbung der Meningen und zu einer gestörten Liquorabsorption durch Entzündung der Arachnoidalzotten. Beide Formen, kommunizierend und nicht kommunizierend, können auftreten 2).
Die granulomatöse Infiltration von Hypothalamus, Hypophysenstiel und Hypophyse beeinträchtigt die endokrine Achse. Bei schwerer Schädigung kann ein zentraler Diabetes insipidus mit Na 168 (Hypernatriämie) auftreten 4).
7. Aktuelle Forschung und Zukunftsperspektiven (Forschungsstadium)
Die MRI-Gefäßwandbildgebungstechnik verspricht eine verbesserte Diagnosegenauigkeit der vaskulitisassoziierten NS.
In einer systematischen Übersichtsarbeit von Focke et al. (2025) gelang der Nachweis von Gefäßwandläsionen mittels MRT-VWI bei 9 von 13 NS-Patienten (69 %) 7). Diese Technik könnte Gefäßwandentzündungen sichtbar machen, die mit der konventionellen Gadolinium-verstärkten MRT schwer zu erfassen sind.
Neopterin und Lysozym im Liquor werden als Biomarker für die vaskulitisassoziierte NS untersucht.
Focke et al. (2025) berichteten, dass bei vaskulitisassoziierter NS das CSF-Neopterin in 100 % der Fälle erhöht war (Mittelwert 5,2 ng/ml) und das Lysozym in 75 % (Mittelwert 4,25 mg/l) 7). Diese Marker könnten zu diagnostischen Werkzeugen werden.
Als Alternative zur FDG-PET werden die Somatostatinrezeptor-Bildgebung und die auf CXCR4 (exprimiert auf aktivierten Makrophagen) abzielende PET-Bildgebung untersucht.
Ach T, Ben Yahia W, Halloul I, Sghaier F, Atig A. Neurosarcoidosis-Induced Hypophysitis Mimicking Pituitary Macroadenoma. Cureus. 2023;15(6):e39865. doi:10.7759/cureus.39865. PMID:37404438; PMCID:PMC10315063.
Kafai Golahmadi A, Craven CL, Watkins LD. Neurosarcoidosis Mimicking Normal Pressure Hydrocephalus. Cureus. 2023;15(6):e40281. doi:10.7759/cureus.40281. PMID:37448383; PMCID:PMC10336621.
Hanif Z, Gonzalez Ramos KN, Razminia P, Aigbe E, Ghafourian P. A Perplexing Case of Bladder Mass Biopsy-Proven Neurosarcoidosis. Cureus. 2023;15(6):e40865. doi:10.7759/cureus.40865. PMID:37489187; PMCID:PMC10363404.
Khawaja MA, Awesat BE, Yasini MN, et al. Neurosarcoidosis Presented as an Isolated Brain Lesion. Cureus. 2023;15(9):e45837. doi:10.7759/cureus.45837.
Chaubey M, Meena K, Singh T, Reddy S, Raj R, Chaudhary A, et al. Neurosarcoidosis: An under-diagnosed cause of myelopathy. Journal of family medicine and primary care. 2024;13(5):2157-2160. doi:10.4103/jfmpc.jfmpc_987_23. PMID:38948561; PMCID:PMC11213433.
Focke JK, Brokbals M, Becker J, Veltkamp R, van de Beek D, Brouwer MC, et al. Cerebral vasculitis related to neurosarcoidosis: a case series and systematic literature review. Journal of neurology. 2025;272(2):135. doi:10.1007/s00415-024-12868-2. PMID:39812656; PMCID:PMC11735521.
Ryan Shields, Olivia Sagan, Logan Roebke, Josh Vander Maten, Shailen Shah, George Chang, Dalia Ibrahim, Sumayya Naz. Rare case of multifocal extradural and intramedullary neurosarcoidosis without pulmonary involvement: a case report and literature review. Spinal Cord Ser Cases. 2021;7(1). doi:10.1038/s41394-021-00450-1.
Sarac E, Erzurum SA, Arif A. An Unusual Presentation of Neurosarcoidosis. Am J Case Rep. 2022;23:e937125. doi:10.12659/ajcr.937125.
Desmond PK, Ben JB, Elizabeth MG, Gordon TP. Optic neuropathy associated with systemic sarcoidosis. Neurol Neuroimmunol Neuroinflammation. 2016;3:e270.
Kidd D, Beynon HL.. The neurological complications of systemic sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis. 2003;20(2):85-94. PMID:12870717.
Kopieren Sie den Artikeltext und fügen Sie ihn in den KI-Assistenten Ihrer Wahl ein.
Artikel in die Zwischenablage kopiert
Öffnen Sie unten einen KI-Assistenten und fügen Sie den kopierten Text in den Chat ein.