El síndrome endotelial iridocorneal (síndrome ICE) es una enfermedad en la que las células endoteliales corneales anormales se extienden y contraen hacia el ángulo y el iris con un tejido membranoso, obstruyendo la malla trabecular y causando elevación de la presión intraocular. Se caracteriza por edema corneal debido a la anomalía endotelial corneal, deformación del iris por contracción del tejido membranoso y sinequias anteriores periféricas (PAS).
El síndrome ICE se clasifica en los siguientes tres tipos clínicos.
Síndrome de Chandler: el más común (aproximadamente 50%). El edema corneal es predominante, con hallazgos iridianos leves.
Atrofia progresiva del iris: formación de agujeros en el iris, desviación pupilar y ectropión uveal son prominentes. Es la que más frecuentemente se asocia con glaucoma.
Síndrome de Cogan-Reese: caracterizado por nódulos pigmentados en la superficie del iris. El subtipo más raro 4).
Generalmente ocurre de forma unilateral en mujeres de 20 a 50 años 2). Es esporádico y no tiene una asociación consistente con otras enfermedades oculares o sistémicas. El síndrome ICE es unilateral y esporádico, lo que constituye el punto de diferenciación más simple con la distrofia corneal polimorfa posterior (PPMD), que es bilateral y hereditaria.
Q¿Cómo se diferencian los tres tipos de síndrome ICE?
A
La diferenciación de los tres tipos se basa principalmente en los hallazgos clínicos del iris y la córnea. El síndrome de Chandler se caracteriza por edema corneal predominante con cambios iridianos mínimos, y el edema corneal puede ocurrir incluso con presión intraocular normal. La atrofia progresiva del iris presenta policoria, desviación pupilar, formación de agujeros en el iris y ectropión uveal, y es la que más frecuentemente se asocia con glaucoma. El síndrome de Cogan-Reese se caracteriza por nódulos marrones pediculados o lesiones pigmentarias difusas en la superficie del iris, y generalmente no se observa atrofia del iris. Sin embargo, todos los tipos comparten una fisiopatología básica común y un enfoque de tratamiento similar.
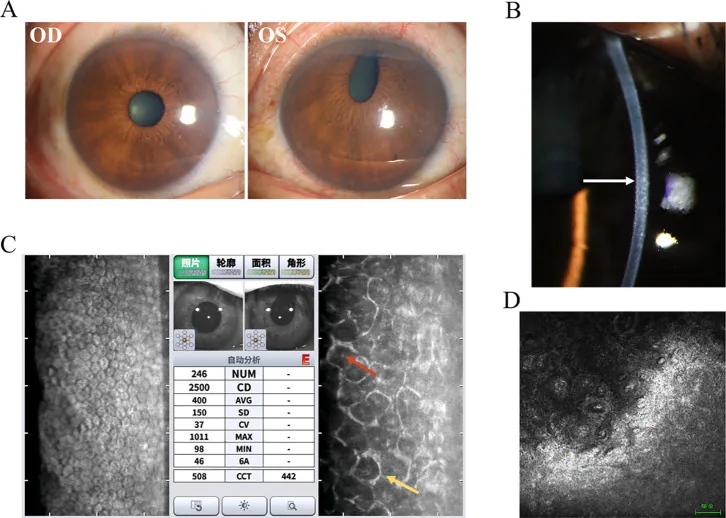
Ma H, et al. Iridocorneal endothelial syndrome. Front Ophthalmol (Lausanne). 2025. Figure 1. PMCID: PMC12537377. License: CC BY.
Imagen clínica y hallazgos de pruebas complementarias del síndrome de Chandler: (A) pérdida del iris y distorsión pupilar, (B) aspecto de plata martillada del endotelio corneal, (C) células ICE en microscopía especular, (D) células endoteliales epitelioides en microscopía confocal. Corresponde a las anomalías del endotelio corneal tratadas en la sección “2. Síntomas principales y hallazgos clínicos”.
Los síntomas iniciales son dolor unilateral, visión borrosa o cambios en la apariencia del iris. El dolor debido al edema corneal y el dolor por el aumento de la presión intraocular debido al cierre angular pueden coexistir. Algunos pacientes consultan tras notar heterocromía del iris o deformación pupilar.
Hallazgos corneales: El endotelio corneal tiene una apariencia de “plata batida” o “cobre martillado”, similar a las gotas de la distrofia endotelial de Fuchs. El edema corneal puede ser microquístico incluso con presión intraocular normal.
Hallazgos del iris: Heterocromía del iris, ectropión uveal, desviación pupilar, formación de agujeros en el iris y atrofia del iris aparecen según el subtipo 2). En el síndrome de Cogan-Reese se observan nódulos pigmentados en la superficie del iris, que histológicamente son lesiones benignas compuestas por células névicas fusiformes que contienen melanina 4).
Hallazgos angulares: La gonioscopia revela sinequias anteriores periféricas que se extienden más allá de la línea de Schwalbe, lo cual es patognomónico del síndrome ICE5). Las sinequias anteriores periféricas son parcheadas y están ubicadas muy anteriormente, y la malla trabecular entre las sinequias parece normal 5).
La verdadera etiología del síndrome ICE sigue sin conocerse, pero existe la hipótesis de que la infección latente por el virus del herpes simple (VHS) o el virus de Epstein-Barr (VEB) induce una inflamación de bajo grado a nivel del endotelio corneal, lo que lleva a una activación similar a la epitelial. Las pruebas de PCR han detectado ADN del VHS en altas tasas en el endotelio corneal y el humor acuoso de pacientes con síndrome ICE, y múltiples informes detectan ADN del VHS en más del 60% de las muestras de córnea y humor acuoso de los pacientes 2).
Patológicamente, las células endoteliales normales son reemplazadas por células similares a las epiteliales con propiedades migratorias. La microscopía electrónica revela características epiteliales como desmosomas, tonofilamentos y microvellosidades. También se ha informado daño tóxico (cambios necróticos) a las células endoteliales normales adyacentes.
Microscopía especular: La herramienta diagnóstica más importante 2). Se pierde la forma hexagonal típica del endotelio corneal, mostrando pleomorfismo y “inversión claro-oscuro”. Son características las células endoteliales más grandes y oscuras (células ICE) con un punto central brillante. Se observa una marcada disminución de la densidad de células endoteliales corneales, y se ha informado de un caso de síndrome de Cogan-Reese que disminuyó a 763 células/mm² 3).
Microscopía confocal in vivo (IVCM): Representa células endoteliales corneales hinchadas en forma de adoquín, pérdida de la estructura hexagonal, células pleomórficas hiperreflectivas y células endoteliales gigantes mononucleares y binucleares 2).
OCT de segmento anterior (AS-OCT): Visualiza adherencias del ángulo iridocorneal, engrosamiento hiperreflectivo de la capa endotelial corneal y tejido membranoso sobre el iris2).
Gonioscopía: Esencial para evaluar las sinequias anteriores periféricas, utilizada para el diagnóstico y seguimiento del glaucoma secundario de ángulo cerrado.
Evaluación del glaucoma: La medición de la presión intraocular, la fotografía del disco óptico, la prueba del campo visual (Humphrey o Goldmann) y la evaluación de la capa de fibras nerviosas de la retina mediante OCT deben incluirse en la evaluación inicial y el seguimiento 2).
Q¿Por qué el síndrome ICE a veces se diagnostica erróneamente como glaucoma de ángulo abierto?
A
La membrana de células endoteliales corneales en avance puede cerrar funcionalmente la malla trabecular sin contracción. En estos casos, la gonioscopía puede no revelar sinequias anteriores periféricas evidentes, lo que lleva a un diagnóstico erróneo de glaucoma de ángulo abierto a pesar de la presencia de un cierre angular “funcional”. Cuando se encuentra glaucoma unilateral, se debe considerar el síndrome ICE en el diagnóstico diferencial y evaluar cuidadosamente el endotelio corneal con microscopía especular.
Supresores de la producción de humor acuoso: Los betabloqueantes tópicos, los agonistas alfa y los inhibidores de la anhidrasa carbónica son tratamientos de primera línea 6). Se recomiendan los supresores de la producción de humor acuoso sobre los mióticos que actúan sobre la vía de drenaje del humor acuoso.
Fármacos relacionados con prostaglandinas: Debido al papel teórico del VHS, existe preocupación por la recurrencia del VHS inducida por latanoprost, por lo que se debe considerar cuidadosamente 2).
Manejo del edema corneal: Se realiza deshidratación corneal con gotas o gel de solución salina hipertónica.
Pronóstico a largo plazo: El tratamiento farmacológico a menudo se vuelve resistente debido a la progresión del tejido membranoso y la expansión de las sinequias anteriores periféricas 6).
Tratamiento Quirúrgico
Trabeculectomía: Se realiza con agentes antifibróticos adyuvantes (MMC o 5-FU) 6). Las tasas de supervivencia reportadas son 73% al año, 44% a los 3 años y 29% a los 5 años. Existe riesgo de oclusión de la fístula por endotelio anormal.
Cirugía de derivación con tubo: Las tasas de supervivencia son 71% al año, 71% a los 3 años y 53% a los 5 años, con mejores resultados a largo plazo que la trabeculectomía5)6). A menudo se eligen derivaciones con placa.
Trasplante de endotelio corneal: DSAEK/DSEK mejora la función corneal 1). La supervivencia del injerto es comparable a la queratoplastia penetrante (PKP), pero la recuperación visual es más rápida y el astigmatismo es menor 1).
Procedimientos ciclodestructivos: Se realiza fotocoagulación con láser de diodo del cuerpo ciliar como último recurso en casos refractarios 6).
Se ha reportado un enfoque quirúrgico en dos etapas: primero, facoemulsificación con implante de iris artificial, seguido de DSAEK 6 meses después 1). En el caso de una mujer de 54 años, la agudeza visual corregida postoperatoria mejoró de 20/100 a 20/32, y se mantuvo una densidad de células endoteliales corneales de 1,640 células/mm² 1).
Se ha reportado una rara complicación de edema macular quístico (EMQ) en el síndrome de Cogan-Reese 3). Se resolvió con AINE tópicos (flurbiprofeno 3 veces al día) pero recurrió tras la suspensión 3). Se hipotetiza que las células endoteliales anormales alteran la barrera hematorretiniana interna, y la producción de prostaglandinas a través de citocinas relacionadas con el VHS está involucrada en la patogenia 3).
La anormalidad fundamental en el síndrome ICE es la metaplasia epitelial del endotelio corneal. Las células endoteliales normales son reemplazadas por células similares a epiteliales con capacidad proliferativa y migratoria. Este proceso también es común en la PPMD, pero el ICE es unilateral y esporádico, mientras que la PPMD es bilateral y autosómica dominante.
Las células endoteliales metaplásicas epiteliales avanzan desde la superficie posterior de la córnea a través de la línea de Schwalbe sobre la malla trabecular y luego hacia la superficie anterior del iris5). La contracción de este tejido membranoso produce las siguientes condiciones.
Glaucoma secundario de ángulo cerrado: El mecanismo principal es la obstrucción del flujo de salida del humor acuoso debido a la formación de sinequias anteriores periféricas extensas 5). El cierre funcional de la malla trabecular por la membrana misma también puede ocurrir, por lo que el glaucoma puede desarrollarse incluso sin sinequias anteriores periféricas evidentes.
Edema corneal: Causado tanto por la disfunción de la bomba de las células endoteliales degeneradas como por la elevación de la presión intraocular debida al glaucoma. Es más prominente en el síndrome de Chandler.
Deformación del iris: La contracción del tejido membranoso tira del iris, resultando en corectopia, formación de agujeros en el iris y ectropión uveal. Es más prominente en la atrofia progresiva del iris, y la corectopia se dirige hacia el área con más sinequias anteriores periféricas.
Los nódulos del iris en el síndrome de Cogan-Reese están compuestos histológicamente por células fusiformes similares a nevos melanocíticos, con una tasa de positividad de Ki-67 menor del 1% y positividad para melan-A, lo que indica lesiones benignas 4). Raramente se asocian con fragilidad zonular, y puede observarse diálisis zonular difusa durante la cirugía de cataratas 4).
Q¿Cómo diferenciar el síndrome ICE de la distrofia corneal polimorfa posterior (PPMD)?
A
El punto de diferenciación más simple es que el síndrome ICE es esporádico y unilateral, mientras que la PPMD es autosómica dominante y bilateral. La microscopía especular también difiere: el síndrome ICE muestra áreas oscuras con un punto brillante central (células ICE), mientras que la PPMD presenta vesículas típicas o estructuras en bandas. La PPMD también puede presentar corectopia y edema corneal, pero es congénita y sin diferencia de género, por lo que el curso clínico es diferente.
Pinheiro-Costa J, Maia J, Branco A, et al. Two-step iridocorneal endothelial syndrome management: endocapsular intraocular lens implantation and Descemet’s stripping automated endothelial keratoplasty. Case Rep Ophthalmol. 2023;14(1):478-486.
Guler Canozer D, Unlu M, Gultekin Irez B, Ozkurt Y. In vivo confocal microscopy and anterior segment optical coherence tomography findings in iridocorneal endothelial syndrome. Turk J Ophthalmol. 2024;54(5):325-330.
Bouvarel H, Hamard P, Agard E, Billant J, El Chehab H, Dot C. Macular edema in Cogan-Reese syndrome. American journal of ophthalmology case reports. 2022;25:101318. doi:10.1016/j.ajoc.2022.101318. PMID:35128161; PMCID:PMC8810354.
Chhadva P, Del Valle Estopinal M, Farid M. IrisNevus (Cogan-Reese) Syndrome Presenting with Zonular Dehiscence during Cataract Extraction. Case reports in ophthalmology. 2022;13(2):435-440. doi:10.1159/000524823. PMID:35950024; PMCID:PMC9247537.
Pazos M, Traverso CE, Viswanathan A; European Glaucoma Society. European Glaucoma Society - Terminology and guidelines for glaucoma, 6th Edition. Br J Ophthalmol. 2025;109(Suppl 1):1-212. doi:10.1136/bjophthalmol-2025-egsguidelines. PMID:41026937.