Das Aniridie-Fibrose-Syndrom (AFS) ist eine seltene Komplikation nach intraokularen Eingriffen bei Patienten mit kongenitaler Aniridie1).
Aus dem rudimentären Irisstumpf bildet sich eine fibröse Membran, die auf die Intraokularlinse, den Ziliarkörper und die vordere Netzhaut übergreifen kann. Erstbeschrieben wurde es 2005 von Tsai et al. an 7 Augen von 6 Patienten als „progressives Vorderabschnittsfibrose-Syndrom nach Operation bei kongenitaler Aniridie“1). Zum Zeitpunkt des Fallberichts von Banifatemi et al. (2024) waren weltweit nur 19 Fälle dokumentiert1).
Die kongenitale Aniridie ist eine panophthalmale Erkrankung, die durch eine partielle bis vollständige Irisaplasie gekennzeichnet ist1).
Die Inzidenz liegt bei 1:64.000 bis 1:96.0001). 90% der Fälle beruhen auf Mutationen des PAX6-Gens (11p13); der Erbgang ist autosomal-dominant mit hoher Penetranz1). Zwei Drittel der Fälle sind familiär, ein Drittel sporadisch1).
Bei angeborener Aniridie treten neben dem Irisdefekt auch verschiedene Augenkomplikationen auf.
Die Sehschärfe ist in der Regel auf 20/100 bis 20/200 eingeschränkt1). Aufgrund der Fragilität der Zonulafasern ist die Implantation einer Intraokularlinse nur mit Vorsicht indiziert.
Das PAX6-Genprodukt liegt auf Chromosom 11p13 in unmittelbarer Nähe des Tumorsuppressorgens WT1, was als contiguous gene syndrome zum WAGR-Syndrom (Wilms-Tumor, Aniridie, urogenitale Fehlbildungen, geistige Behinderung) führen kann.
QTritt bei Patienten mit kongenitaler Aniridie nach intraokularen Eingriffen zwangsläufig das Aniridie-Fibrose-Syndrom auf?
A
Das Aniridie-Fibrose-Syndrom tritt nach intraokularen Eingriffen auf, ist jedoch eine seltene Komplikation, die nicht bei allen Patienten auftritt. Eine systematische Übersicht zur Implantation künstlicher Iris berichtet eine Inzidenz von 3,1 %1); der genaue Pathomechanismus ist noch nicht vollständig geklärt.
Ni W, et al. A novel histopathologic finding in the Descemet’s membrane of a patient with Peters Anomaly: a case-report and literature review. BMC Ophthalmol. 2015. Figure 2. PMCID: PMC4619091. License: CC BY.
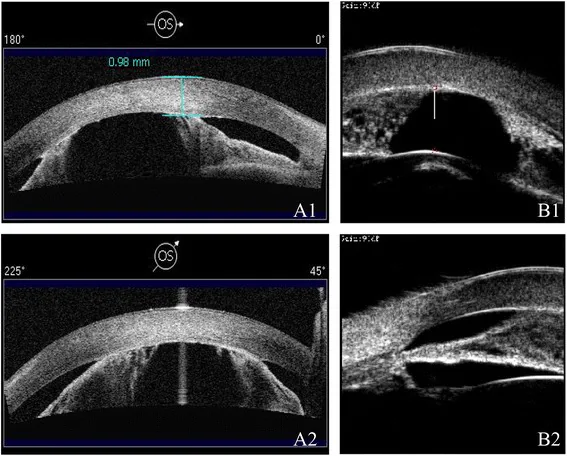
AS-OCT (a) und UBM (b) des linken Auges zeigen eine flache Vorderkammer und periphere anteriore Synechien. Dies entspricht den im Abschnitt „2. Hauptsymptome und klinische Befunde“ behandelten anterioren Synechien.
Das Hauptsymptom des Aniridie-Fibrose-Syndroms ist eine schmerzlose, allmähliche Sehverschlechterung gegenüber dem Ausgangswert1). Da keine Schmerzen auftreten, wird die Vorstellung beim Arzt oft verzögert. In fortgeschrittenen Stadien können aufmerksame Patienten das Vorhandensein einer Membran bemerken.
Vom Iriswurzel ausgehende fibröse Membran: Im Spaltlampenmikroskop kann eine Membran sichtbar sein, die von der rudimentären Iriswurzel gebildet wird. Sie betrifft häufig die Intraokularlinse.
Anteriorverlagerung der Intraokularlinse: Durch den Zug der Membran bewegt sich die Intraokularlinse nach vorne und kann bei Fortschreiten die Hornhaut berühren.
Befunde im fortgeschrittenen Stadium
Hypotonie: Entsteht, wenn die Membran den Ziliarkörper bedeckt. Im von Banifatemi et al. berichteten Fall wurde ein IOD von 0 mmHg im rechten Auge gemessen1).
Hornhautendothelinsuffizienz: Führt schließlich zu einem generalisierten Hornhautödem und Endothelschaden. Im selben Fallbericht wurde ein totales Hornhautödem festgestellt1).
Netzhautablösung: Kann auftreten, wenn sich die Membran auf die vordere Netzhaut ausdehnt.
Im von Banifatemi et al. berichteten Fall (3-jähriges Mädchen, 2 Jahre nach bilateraler Ahmed-Glaukomventil-Operation) wurden am rechten Auge folgende Befunde erhoben1):
Hypotonie (IOD 0 mmHg) mit totalem Hornhautödem
Nach oben subluxierte reife Katarakt und inferiore Zonulolyse
Weiße vaskularisierte Membran, die von der rudimentären Iris bis zum unteren Teil der subluxierten Linse reicht
Ultraschallbiomikroskopie: Dicke fibröse Membran, die von der Iriswurzel ausgeht und sich bis zum hinteren Teil der kataraktösen Linse erstreckt
B-Scan: Axiale Länge 21 mm, Papillenödem, flache Aderhautablösung im vorderen Zweidrittel (keine Netzhautablösung)
QWarum treten Hypotonie und Hornhautödem auf?
A
Die Hypotonie entsteht, weil die fibröse Membran den Ziliarkörper bedeckt und dessen Funktion beeinträchtigt, was zu einer verminderten Kammerwasserproduktion führt1). Das Hornhautödem wird durch den Kontakt der Intraokularlinse mit der Hornhaut oder durch eine Hornhautendothelschädigung infolge der Hypotonie verursacht.
Die Ätiologie des Aniridie-Fibrose-Syndroms ist unbekannt. Alle berichteten Fälle traten jedoch nach intraokularen Eingriffen auf.
Zu den Risikofaktoren gehören:
Intraokulare Operationen in der Vorgeschichte: Am häufigsten Kataraktoperation mit Intraokularlinsenimplantation
Einlage intraokularer Vorrichtungen: Mehrere intraokulare Geräte wie Glaukom-Drainage-Implantate (Tube-Shunts) sind beteiligt
Weibliches Geschlecht: 88 % der berichteten Fälle waren weiblich
Mehrere intraokulare Operationen: Im ersten Bericht von Tsai et al. hatten alle Fälle mehrere intraokulare Operationen in der Vorgeschichte1)
Operationsvorgeschichte von 7 Augen bei 6 Patienten nach Tsai et al.1): Kataraktoperation mit Hinterkammerlinse bei 7 Augen, Tube-Shunt bei 6 Augen, Hornhauttransplantation bei 4 Augen. 9 Fälle von Bakhtiari et al.1): Kataraktoperation mit Intraokularlinsenimplantation bei allen, Tube-Shunt bei 7, Hornhauttransplantation/limbale Allotransplantation bei 7.
Zwei Theorien zum Entstehungsmechanismus wurden vorgeschlagen.
Mechanische Reiztheorie: Intraokulare Vorrichtungen berühren rudimentäres Irisgewebe oder unreife Irisgefäße und bieten so ein Gerüst für die Membranbildung1)
PAX6-Mutations-assoziierte Fibrose-Prädispositionstheorie: PAX6 reguliert den Wnt-Signalweg negativ; eine chronische Überaktivierung des Wnt-Signalwegs durch PAX6-Mutationen fördert die Fibrose. Mausmodellstudien von Wang et al. bestätigten, dass eine PAX6-Haploinsuffizienz bereits vor der Operation einen präfibrotischen Zustand induziert1)
Bemerkenswerterweise wurde auch bei einem 8 Monate alten Kind nach Descemet-Membran-Endothel-Keratoplastik (DSAEK) eine rezidivierende progressive Fibrose berichtet1), was darauf hindeutet, dass die Erkrankung auch ohne intraokulare Vorrichtungen auftreten kann.
Die Diagnose des Aniridie-Fibrose-Syndroms erfolgt durch klinische Beobachtung bei Patienten mit angeborener Aniridie und intraokularen Operationen in der Vorgeschichte.
Die regelmäßige Spaltlampenuntersuchung ist die Grundlage der Diagnose 1). Dabei kann eine fibröse Membran sichtbar gemacht werden, die von der Iriswurzel ausgeht und die Intraokularlinse umhüllt. In einem frühen Stadium, wenn die Hornhaut noch klar ist, ist eine detaillierte Beurteilung des vorderen Augenabschnitts möglich.
Diese Untersuchung ist unverzichtbar, wenn Trübungen der vorderen Augenmedien wie Hornhauttrübungen vorliegen 1).
Beurteilung des Zustands des Ziliarkörpers
Erfassung der Ausdehnung der fibrösen Membran
Bestätigung der Bildung einer zyklitischen Membran
Im Fallbericht von Banifatemi et al. wurde die Ultraschallbiomikroskopie unter Chlorhydrat-Anästhesie durchgeführt, wobei eine dicke fibröse Membran von der Iriswurzel bis zur Rückseite der Kataraktlinse bestätigt wurde 1).
Postoperative Entzündungsreaktion: Abgrenzung von einer einfachen postoperativen Entzündungsreaktion (das Aniridie-Fibrose-Syndrom ist durch das Fehlen von Entzündungszeichen gekennzeichnet)
Die Behandlung des Aniridie-Fibrose-Syndroms erfolgt hauptsächlich chirurgisch. Eine frühzeitige Diagnose und Intervention verbessern die Sehprognose 1).
Um ein weiteres Membranwachstum und Gewebezerstörung zu verhindern, wird eine frühe chirurgische Membranektomie mittels perforierender Keratoplastik (PKP) empfohlen.
Von 7 Augen bei 6 Patienten von Tsai et al. wurden 5 operiert 1):
Durchführung einer perforierenden Keratoplastik und Membranektomie
Bei hypotonen Augen erholte sich der Augeninnendruck nach Membranektomie (stabil bei 5–10 mmHg)
Bei allen 5 Fällen wurde eine Sehverbesserung festgestellt
Rezidivrate: Rezidive traten nur bei Patienten mit Explantation/Austausch der Intraokularlinse auf. Gleichzeitige Linsenentfernung wird als wirksam zur Rezidivprävention angesehen.
Sehergebnisse: Bei allen 5 operierten Patienten wurde eine Sehverbesserung festgestellt 1).
Bakhtiari et al. (9 Fälle)
Operationsmethode: Bei allen Patienten wurde eine Boston-KPro Typ 1 implantiert (primär oder sekundär). Bei 7 von 9 Fällen wurde die Intraokularlinse explantiert, bei 8 Fällen eine Vitrektomie durchgeführt.
Die Boston-Keratoprothese Typ 1 ist eine praktikable Behandlungsoption, bei der kein Zusammenhang mit einem Wiederauftreten des Aniridie-Fibrose-Syndroms festgestellt wurde1). Da die KPro entfernt von der Iriswurzel (dem Ausgangspunkt der Läsion) platziert wird, wird angenommen, dass die Reizung der Iris geringer ist.
Es ist jedoch zu beachten, dass bei aniriden Augen eine hintere KPro-Membranbildung mit hoher Rate (61–66 %) auftritt. Diese Rate ist signifikant höher als bei nicht-aniriden Augen (26,7–39 %)1), und diese hintere KPro-Membran könnte ebenfalls ein Phänotyp des Aniridie-Fibrose-Syndroms sein1).
Die Erhaltungsrate der Boston KPro Typ 1 bei aniriden Augen betrug laut Dyer et al. 83,3 % (mittlere Nachbeobachtung 58,7 Monate) und laut Shah et al. 87 % (54 Monate Nachbeobachtung)1).
Bakhtiari et al. empfehlen zur Rezidivprophylaxe nicht nur eine vordere Vitrektomie, sondern eine komplette Vitrektomie1).
QWie ist die Sehprognose ohne Operation?
A
Die Daten zum Verlauf ohne Operation bei gesicherter AFS-Diagnose sind begrenzt. Im Fallbericht von Banifatemi et al. (2024) wurde aufgrund der Ablehnung der Eltern keine Operation durchgeführt1). Allgemein wird bei fehlender Behandlung ein Fortschreiten von Hornhautendothelinsuffizienz, Hypotonie und Netzhautablösung befürchtet. Im Bericht von Tsai et al. zeigte sich bei allen operierten Fällen eine Sehverbesserung, was die Bedeutung einer frühzeitigen Intervention unterstreicht1).
6. Pathophysiologie und detaillierter Pathomechanismus
Der genaue Pathomechanismus des Aniridie-Fibrose-Syndroms ist noch ungeklärt, aber mehrere Hinweise deuten darauf hin, dass die Iriswurzel der Ausgangspunkt der Fibrose ist.
Immunhistochemie: In 2 Fällen wenige T-Zellen und Makrophagen → deutet darauf hin, dass Entzündung nicht die Hauptursache ist (in 1 Fall waren Entzündungszellen vorhanden)
Elektronenmikroskopie: Gemisch aus unreifen Kollagenbündeln und reifen Fasern. Keine Gliazellen, Hornhautendothelzellen oder Linsenepithelzellen
Banifatemi et al. (2024) stellen fest: „Die fibröse Membran ähnelt der postoperativen Membran um die Intraokularlinse bei chronischer Uveitis, unterscheidet sich jedoch dadurch, dass sie ohne Entzündung einhergeht“1).
Die Theorie besagt, dass der Kontakt des Intraokulargeräts mit rudimentärem Irisgewebe oder unreifen Irisgefäßen ein Gerüst für die Membranbildung bietet 1). Dies stimmt mit der klinischen Tatsache überein, dass das Aniridie-Fibrose-Syndrom in Verbindung mit verschiedenen Intraokulargeräten wie Intraokularlinsen, Tubenshunts und künstlichen Iriden auftritt.
PAX6 ist ein Transkriptionsfaktor, der den Wnt-Signalweg negativ reguliert. Eine Theorie besagt, dass eine chronische Überaktivierung des Wnt-Signalwegs durch PAX6-Mutationen eine Fibrose-Prädisposition im Auge bildet 1). Wang et al. bestätigten in Mausmodellstudien, dass PAX6-Haploinsuffizienz (haploPAX6) auch in nicht operierten Augen im Vergleich zum Wildtyp einen präfibrotischen Zustand induziert 1).
Ähnliche progressive Fibrosierung wurde auch bei anderen angeborenen Vorderabschnittsanomalien wie der Peters-Anomalie berichtet, was darauf hindeutet, dass PAX6-Mutationen allgemein mit einer Fibrosierungsneigung assoziiert sind 1).
7. Aktuelle Forschung und zukünftige Perspektiven (Berichte aus der Forschungsphase)
Die Rate der retrospektiven KPro-Membranbildung nach Implantation eines Boston-KPro-Typ-1 bei aniriden Augen beträgt 61–66 %, was deutlich höher ist als bei nicht-aniriden Augen (26,7–39 %) 1).
Yang et al. weisen darauf hin, dass die hohe Rate der post-KPro-Membranbildung ein Phänotyp des Aniridie-Fibrose-Syndroms sein könnte1). Muzychuk et al. berichteten hingegen, dass Aniridie ein wichtiger Risikofaktor für den Sehverlust nach einer Boston-KPro-Operation ist1).
Implantation einer künstlichen Iris und Aniridie-Fibrose-Syndrom
In einer systematischen Übersichtsarbeit zur künstlichen Irisimplantation von Romano et al. wurde berichtet, dass in der Studie von Figueredo und Snyder mit 96 Augen mit Aniridie die Inzidenz des Aniridie-Fibrose-Syndroms 3,1 % betrug 1). Da die künstliche Irisimplantation wie Intraokularlinsen und Tubus-Shunts als intraokulares Gerät fungiert, birgt sie ein potenzielles Risiko für die Entwicklung des Aniridie-Fibrose-Syndroms.
Molekulare Mechanismen des Aniridie-Fibrose-Syndroms
Die Mausmodellstudie von Wang et al. ist eine der ersten systematischen Untersuchungen zur Aufklärung der molekularen Mechanismen des Aniridie-Fibrose-Syndroms 1). Es wird angedeutet, dass die selektive Hemmung des Wnt-Signalwegs ein zukünftiges therapeutisches Ziel darstellen könnte, jedoch ist es noch nicht zur klinischen Anwendung gekommen.
Banifatemi M, Razeghinejad R, Salouti R, Abolfathzadeh N. Aniridic fibrosis syndrome in a child with Ahmed glaucoma valve: Report of a case and review of the literature. J Curr Ophthalmol. 2024;36:453-6. doi:10.4103/joco.joco_155_24.
Bakhtiari P, Chan C, Welder JD, de la Cruz J, Holland EJ, Djalilian AR. Surgical and visual outcomes of the type I Boston Keratoprosthesis for the management of aniridic fibrosis syndrome in congenital aniridia. Am J Ophthalmol. 2012;153(5):967-971.e2. PMID: 22265154.
Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, et al. PAX6-Related Aniridia. . 1993. PMID: 20301534.
Kopieren Sie den Artikeltext und fügen Sie ihn in den KI-Assistenten Ihrer Wahl ein.
Artikel in die Zwischenablage kopiert
Öffnen Sie unten einen KI-Assistenten und fügen Sie den kopierten Text in den Chat ein.