急性期

Harding病
一目了然的要點
Section titled “一目了然的要點”1. 什麼是Harding病?
Section titled “1. 什麼是Harding病?”Harding病是一種在Leber遺傳性視神經病變(LHON)的粒線體DNA(mtDNA)突變背景下,共存類似多發性硬化症(MS)的脫髓鞘神經症狀的疾病。1992年,Harding等人報告了8例有LHON家族史的雙側視神經病變女性患者(其中6例有符合MS的神經症狀),該病因此得名。1)
文獻中迄今共報告了88例。1)患者中70.4%(62例)為女性,男女比例為2.38:1。1)這與LHON多見於男性(男性佔93.1%)形成對比,提示MS的女性優勢易感性可能參與其中。平均發病年齡為30.5歲。1)
LHON的盛行率在日本約為1/50,000(全國總患者數估計約4,000~5,000人),英國為1/31,000,澳洲為1/68,000。1)2015年,LHON在日本被指定為難病,並確立了診斷標準。每年新發病例約117人,其中30歲前發病佔47%。
Q
Harding病有多罕見?
2. 主要症狀與臨床所見
Section titled “2. 主要症狀與臨床所見”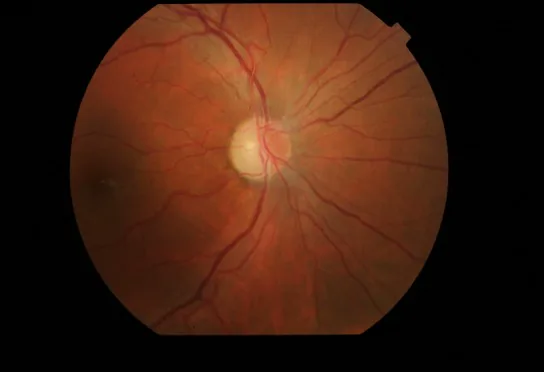
Cristina Culea, Bogdana Tăbăcaru, Simona Stanca et al. Leber’s Hereditary Optic Neuropathy – Case Discussion. Romanian Journal of Ophthalmology. 2019 Jan-Mar; 63(1):91. Figure 5. PMCID: PMC6531774. License: CC BY.
顯示Harding病的圖片
- 從霧視開始的視力下降:從無痛性霧視開始,逐漸惡化至嚴重視力下降和中心暗點。
- 雙眼受累:通常累及雙眼。第二隻眼受累的間隔比LHON長,平均為1.66年。1)
- 多次視力障礙發作:與通常只有兩次發作的LHON不同,Harding病引起多次視力下降發作。1)
- 無眼痛:與MS的視神經炎不同,不伴隨眼痛。1)這是鑑別診斷的重要要點。
- 眼外症狀:可能伴隨姿勢性震顫、周邊神經病變、運動障礙、心律不整、肌無力、肌肉疾病。
臨床所見(醫師檢查確認的發現)
Section titled “臨床所見(醫師檢查確認的發現)”慢性期
視神經盤萎縮:炎症消退後殘留視神經盤萎縮。
RNFL變薄:OCT顯示以乳頭黃斑束為主的視網膜內層變薄。
最佳矯正視力下降:常停留在0.01左右。光覺保留。
瞳孔對光反射保留:與其他視神經疾病相比,瞳孔對光反射得以保留或僅輕微受損。
- 視野:表現為緻密的中心暗點或中心盲點性暗點(centrocecal scotoma)。
Q
Harding病的視力下降與LHON或多發性硬化視神經炎有何不同?
3. 病因與危險因子
Section titled “3. 病因與危險因子”主要mtDNA突變
Section titled “主要mtDNA突變”Harding病是由粒線體DNA(mtDNA)的致病性突變引起的。三種主要突變均編碼複合體I亞基,透過ATP合成障礙和活性氧(ROS)增加誘導視網膜神經節細胞(RGC)凋亡。3)
88例Harding病中突變的分布如下所示。
| 突變 | 基因 | Harding病中的比例 |
|---|---|---|
| m.11778G>A | MT-ND4 | 69.3%(61/88例) |
| m.14484T>C | MT-ND6 | 12.5% |
| m.3460G>A | MT-ND1 | 10.2% |
引用:Alorainy J et al. 20241)
MT-ND4突變在亞洲佔LHON病例的90%,在歐洲佔70%。3)在日本患者中,這三種基因突變也佔95%。
mtDNA從母親傳給子女(母系遺傳)。男性患者的後代不會遺傳。
LHON與MS共存的病理生理假說
Section titled “LHON與MS共存的病理生理假說”- MS修飾LHON假說:mtDNA突變修飾MS的表型,導致無痛性、更嚴重且不可逆的非典型視神經炎。
- MS易感性導致LHON顯現假說:女性中MS易感的遺傳和環境因素,促進無症狀mtDNA突變攜帶者發生LHON。
- 偶發性炎症誘導假說:攜帶LHON mtDNA突變的患者,前部視路被誘導出偶發性炎症反應。
環境風險因素
Section titled “環境風險因素”- 吸菸:可能作為LHON發病的風險因素。
- 大量飲酒:同樣可能參與發病風險。
- 抗結核藥物(如乙胺丁醇等):臨床上有關聯性被指出。
Q
為什麼Harding病在女性中較常見?
4. 診斷與檢查方法
Section titled “4. 診斷與檢查方法”診斷定義與日本診斷標準
Section titled “診斷定義與日本診斷標準”Harding病定義為「符合MS診斷標準且具有主要LHON突變的患者」(Pfeffer等人)。
在日本,採用2015年確立的LHON診斷標準。
- 確定病例:滿足主要徵候(急性至亞急性、雙眼、無痛性視力下降和中心暗點 + 急性期至少一項檢眼鏡異常)。
- 確診病例:主要徵候 + 粒線體基因錯義突變 + MRI無球後視神經異常。
- 疑似病例:母系遺傳明確但未檢測到基因突變。
主要檢查方法
Section titled “主要檢查方法”| 檢查 | 主要目的/所見 |
|---|---|
| mtDNA分析/多基因檢測組 | 識別m.3460G>A、m.11778G>A、m.14484T>C |
| MRI | 確認脫髓鞘性T2高信號白質病變 |
| 螢光眼底攝影 | 視神經盤有無染料滲漏(無滲漏為鑑別點) |
| 光學同調斷層掃描(OCT) | 乳頭黃斑束變薄 |
| 心電圖(ECG) | 排除心臟早期興奮症候群 |
- 基因檢測:在日本,可委外檢測三種突變(m.3460、m.11778、m.14484)。若結果為陰性,需轉介至核心機構。
- 外顯子組定序:若未發現mtDNA突變,則檢測核基因突變,如DNAJC30。
- MRI特徵:區分Harding病與MS的發現包括T2病灶訊號強度降低且邊界不清、T1病灶缺乏高訊號、以及腦室周圍高訊號區(與典型的Dawson手指徵模式不同)。1)LHON-MS和MS分別有73%和90%符合McDonald空間播散標準。1)
- 臨界閃爍頻率(CFF)/ 瞳孔對光反射檢查:保持正常或僅輕度下降。
- 篩檢考量:在1,666名MS患者的篩檢中,僅5人LHON突變陽性,因此不建議常規篩檢。建議僅限於有嚴重或雙眼視野缺損或母系家族史的患者進行個案判斷。1)
需要與中毒性視神經病變、體染色體顯性視神經萎縮(ADOA)、隱匿性黃斑營養不良、錐體營養不良、壓迫性視神經病變、視神經脊髓炎譜系疾病(NMOSD)進行鑑別。
5. 標準治療方法
Section titled “5. 標準治療方法”日本國內的治療
Section titled “日本國內的治療”尚無確立的治療方法。以下為目前的應對措施。
- 艾地苯醌(未核准):輔酶Q10衍生物。輔助電子傳遞,可能維持或改善視功能。日本國內也進行了臨床試驗,部分病例報告視功能改善,但為國內未核准藥物。患者可能透過個人進口口服。
- 補充劑:輔酶Q10、B群維生素、維生素C等,由各醫療機構自行決定使用。
- 戒菸指導:由於吸菸與LHON發病風險相關,因此指導患者戒菸。
- 遺傳諮詢:建議早期對可能將mtDNA突變傳給後代的女性進行遺傳諮詢。提供資訊:男性患者不會將突變傳給子女,存在自然恢復的病例,且該病被認定為罕見疾病。
- 低視力照護:對殘留視力障礙的患者提供適當的照護與生活指導。
海外治療報告(補充)
Section titled “海外治療報告(補充)”在尚未確立標準治療的情況下,已嘗試以下治療方法。
- 甲基潑尼松龍靜脈注射:每日1克,連續3天給藥,部分患者報告有輕度至中度的視力改善。1)
- 米托蒽醌:約19.2mg/月靜脈注射有改善視力和神經症狀的報告,但因嚴重副作用使用受限。1)
- 血漿置換/環磷醯胺:部分報告顯示光感和對比敏感度主觀改善,但結果不一致。1)
- 免疫調節藥物:可穩定臨床和MRI表現,但不能阻止視力障礙進展。有報告稱停用那他珠單抗後出現炎症反彈。1)
Q
日本對Harding病採用何種治療?
6. 病理生理學與詳細發病機制
Section titled “6. 病理生理學與詳細發病機制”LHON的基本機轉源於mtDNA突變導致複合體I(粒線體呼吸鏈)次單元功能障礙。電子傳遞障礙導致ATP合成減少,同時活性氧(ROS)累積。這誘導RGC凋亡,導致視神經萎縮。3)
不同突變的視力恢復率有差異,MT-ND4突變的視力恢復率為4%~25%,低於MT-ND1和MT-ND6突變。3)日本最常見的mt11778(MT-ND4)突變,其視功能改善率僅為百分之幾。mt14484(MT-ND6)突變的改善率最高。
LHON在男性中較高的外顯率提示X染色體連鎖核基因的參與。3)
- PRICKLE3(X染色體Xp11.23):調節ATP合成酶(複合體V)的功能。3)
- YARS2突變:損害電子傳遞鏈複合體I、III、IV的功能。3)
- DNAJC30突變(c.152A>G):損害複合體I修復機制,導致體染色體隱性遺傳LHON。3)
在LHON-MS的組織病理學中,T細胞和活化的巨噬細胞/小膠質細胞介導組織損傷。LHON病變內存在發炎細胞是不尋常的,提示早期免疫機制。1)白質變化不僅包括MS樣脫髓鞘,還包括空泡化和瀰漫性髓鞘蒼白。粒線體功能障礙、自體免疫反應和分子擬態被認為是發炎性脫髓鞘的機制。1)
神經學後遺症範圍從輕度障礙到類似復發緩解型MS的病程。
7. 最新研究與未來展望(研究階段報告)
Section titled “7. 最新研究與未來展望(研究階段報告)”基因治療(rAAV2/2-ND4)
Section titled “基因治療(rAAV2/2-ND4)”針對MT-ND4突變的LHON患者,已開發出基因治療(雷納多基因諾帕博韋克;rAAV2/2-ND4)。透過異位基因表現將野生型ND4蛋白引入RGC的粒線體。2)
REVERSE研究(2019年)報告,玻璃體內注射眼的平均BCVA改善為-0.308 LogMAR,未治療眼為-0.259 LogMAR。2)結果提示存在對側眼的免疫學轉移。匯總分析(RESCUE、REVERSE、RESTORE、REFLECT四項試驗)顯示,與對照組相比,改善約21.5個ETDRS字母。雙側給藥比單側給藥效果更大(約12 vs 約8個ETDRS字母),薈萃分析顯示rAAV2/2-ND4比艾地苯醌更有效,兩者均優於自然病程。2)
目前尚未獲得EMA和FDA核准,對Harding病的有效性尚未驗證。雖然預期對LHON成分有效,但對MS病理的影響尚不明確。2)
間質幹細胞(MSC)治療
Section titled “間質幹細胞(MSC)治療”正在研究將iPSC來源的MSC玻璃體內注射,直接將粒線體轉移至RGC的方法。F-肌動蛋白依賴的隧道納米管(TNT)介導轉移。在Ndufs4 KO小鼠中已報告可防止RGC密度降低,但尚未進入臨床試驗。3)
4-氨基吡啶
Section titled “4-氨基吡啶”有研究表明4-氨基吡啶可能改善MS患者的視覺誘發電位(VEP),正在探討其對視力預後不良的Harding病患者的應用。
需要開發針對Harding病的特異性生物標誌物並進行前瞻性研究。對於具有LHON和MS雙重病理的該疾病,治療應個體化,多學科協作至關重要。
Q
基因治療能否用於Harding病?
8. 參考文獻
Section titled “8. 參考文獻”- Alorainy J, Alorfi Y, Karanjia R, Badeeb N. A Comprehensive Review of Leber Hereditary Optic Neuropathy and Its Association with Multiple Sclerosis-Like Phenotypes Known as Harding’s Disease. Eye and brain. 2024;16:17-24. doi:10.2147/EB.S470184. PMID:39100385; PMCID:PMC11296356.
- Wong DCS, Makam R, Yu-Wai-Man P. Advanced therapies for inherited optic neuropathies. Eye (London, England). 2026;40(2):177-184. doi:10.1038/s41433-025-04109-1. PMID:41318849; PMCID:PMC12830909.
- Hu JL, Hsu CC, Hsiao YJ, et al. Leber’s hereditary optic neuropathy: Update on the novel genes and therapeutic options. J Chin Med Assoc. 2024;87:12-16.
- Sanders FWB, Votruba M. Outcomes of idebenone therapy for Leber hereditary optic neuropathy in a cohort of patients from Wales. Eye (Lond). 2025 Sep 17;39(16):2952-2957. doi:10.1038/s41433-025-03993-x. PMID:40962867; PMCID:PMC12583466.