Die Harding-Krankheit ist eine Erkrankung, bei der demyelinisierende neurologische Symptome ähnlich der Multiplen Sklerose (MS) vor dem Hintergrund einer mitochondrialen DNA (mtDNA)-Mutation der Leberschen hereditären Optikusneuropathie (LHON) koexistieren. Sie ist nach dem Bericht von Harding et al. aus dem Jahr 1992 benannt, in dem acht Frauen mit bilateraler Optikusneuropathie und LHON-Familienanamnese (darunter sechs mit MS-konformen neurologischen Symptomen) beschrieben wurden. 1)
In der Literatur wurden bisher insgesamt 88 Fälle berichtet. 1) 70,4 % (62 Fälle) der Patienten sind weiblich, das Geschlechterverhältnis beträgt 2,38:1. 1) Dies steht im Gegensatz zur LHON, die häufiger bei Männern auftritt (93,1 %), und deutet auf eine Beteiligung der weiblichen Prädisposition für MS hin. Das durchschnittliche Erkrankungsalter beträgt 30,5 Jahre. 1)
Die Prävalenz der LHON wird in Japan auf etwa 1/50.000 (geschätzte Gesamtzahl der Patienten im Land etwa 4.000–5.000), im Vereinigten Königreich auf 1/31.000 und in Australien auf 1/68.000 geschätzt. 1) 2015 wurde LHON in Japan als seltene Krankheit eingestuft und diagnostische Kriterien wurden festgelegt. Die Zahl der Neuerkrankungen beträgt etwa 117 pro Jahr, wobei 47 % vor dem 30. Lebensjahr auftreten.
QWie selten ist die Harding-Krankheit?
A
In der Literatur wurden nur 88 Fälle berichtet, was sie zu einer äußerst seltenen Erkrankung macht. 1) 70,4 % der Patienten sind weiblich, das Durchschnittsalter beträgt 30,5 Jahre. Da das gleichzeitige Vorliegen von LHON und MS erforderlich ist, ist sie viel seltener als LHON oder MS allein.
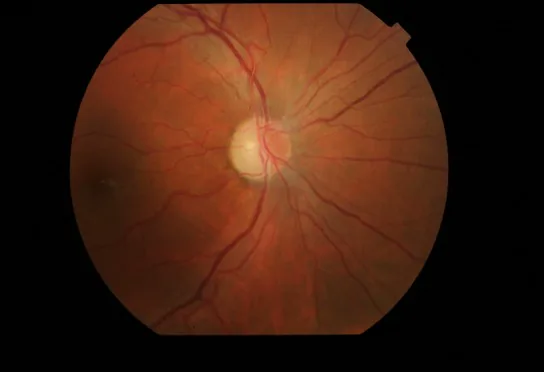
Sehverschlechterung beginnend mit Nebelsehen : beginnt mit schmerzlosem Nebelsehen, verschlechtert sich allmählich und führt zu schwerer Sehverschlechterung und zentralem Skotom.
Beidseitige Störung : betrifft normalerweise beide Augen. Der Abstand bis zur Beteiligung des zweiten Auges ist länger als bei LHON, im Durchschnitt 1,66 Jahre. 1)
Mehrere Episoden von Sehstörungen : Während LHON normalerweise nur zwei Episoden verursacht, führt die Harding-Krankheit zu mehreren Episoden von Sehverschlechterung. 1)
Ohne Augenschmerzen : Im Gegensatz zur Optikusneuritis bei MS tritt keine Augenschmerzen auf. 1) Dies ist ein wichtiger Punkt für die Differenzialdiagnose.
Extraokuläre Symptome : Kann mit Haltetremor, peripherer Neuropathie, Bewegungsstörungen, Herzrhythmusstörungen, Muskelschwäche, Muskelerkrankungen einhergehen.
Klinische Befunde (vom Arzt bei der Untersuchung festgestellte Befunde)
Rötung und Hyperämie der Papille : Mit peripapillärem RNFL-Ödem.
Retinale Teleangiektasien : Erweiterung der Kapillaren (Teleangiektasie) und vermehrte Gefäßschlängelung.
Kein Leck in der Fluoreszenzangiographie : Von der geröteten Papille wird kein Fluoreszenzfarbstoff ausgetreten. Dies ist ein wichtiges Unterscheidungsmerkmal zur Optikusneuritis.
OCT-Befunde : Vor Beginn der Symptome soll eine Schwellung der Papille nachweisbar sein.
Chronische Phase
Papillenatrophie : Nach Abklingen der Entzündung bleibt eine Papillenatrophie bestehen.
RNFL-Verdünnung : Im OCT zeigt sich eine Verdünnung der inneren Netzhautschichten, hauptsächlich im papillomakulären Bündel.
Verminderter bestkorrigierter Visus : Er bleibt oft um 0,01. Die Lichtwahrnehmung bleibt erhalten.
Erhalt des Pupillenlichtreflexes: Im Vergleich zu anderen Sehnervenerkrankungen bleibt der Pupillenlichtreflex erhalten oder ist nur geringfügig beeinträchtigt.
Gesichtsfeld: Zeigt ein dichtes Zentralskotom oder ein zentrozökales Skotom (centrocecal scotoma).
QWie unterscheidet sich die Sehverschlechterung bei der Harding-Krankheit von der bei LHON oder Optikusneuritis bei MS?
A
Der Unterschied zur LHON besteht darin, dass mehrere Episoden von Sehverschlechterung auftreten und der Zeitabstand bis zur Beteiligung des zweiten Auges länger ist (durchschnittlich 1,66 Jahre). 1) Der Unterschied zur Optikusneuritis bei MS ist das Fehlen von Augenschmerzen und das Fehlen von Farbstoffaustritt aus der Papille in der Fluoreszenzangiographie. Diese Merkmale sind wichtige Hinweise für die Differentialdiagnose der Harding-Krankheit.
Die Ursache der Harding-Krankheit sind pathogene Mutationen der mitochondrialen DNA (mtDNA). Die drei Hauptmutationen kodieren alle für Untereinheiten des Komplexes I und induzieren über eine gestörte ATP-Synthese und erhöhte reaktive Sauerstoffspezies (ROS) die Apoptose retinaler Ganglienzellen (RGC). 3)
Die Verteilung der Mutationen bei 88 Fällen der Harding-Krankheit ist unten dargestellt.
Mutation
Gen
Anteil bei Harding-Krankheit
m.11778G>A
MT-ND4
69,3 % (61/88 Fälle)
m.14484T>C
MT-ND6
12,5 %
m.3460G>A
MT-ND1
10,2 %
Zitat: Alorainy J et al. 20241)
Die MT-ND4-Mutation macht in Asien 90 % und in Europa 70 % der LHON-Fälle aus.3) Auch bei japanischen Patienten machen diese drei Genmutationen 95 % der Fälle aus.
mtDNA wird von der Mutter auf das Kind übertragen (mütterliche Vererbung). Bei männlichen Patienten wird sie nicht an die Nachkommen weitergegeben.
Pathologische Hypothese des gleichzeitigen Auftretens von LHON und MS
Für den Mechanismus des gleichzeitigen Auftretens von LHON und MS werden die folgenden drei Hypothesen vorgeschlagen.1)
MS-modifizierte LHON-Hypothese: Die mtDNA-Mutation modifiziert den Phänotyp der MS und verursacht eine schmerzlose, schwerere und irreversible atypische Optikusneuritis.
MS-Prädispositions-Theorie der LHON-Manifestation: Bei Frauen fördern genetische und umweltbedingte Faktoren, die für MS prädisponieren, das Auftreten von LHON bei asymptomatischen Trägern von mtDNA-Mutationen.
Theorie der zufälligen Entzündungsinduktion: Bei Patienten mit LHON-mtDNA-Mutationen wird eine zufällige Entzündungsreaktion im vorderen Sehweg induziert.
Rauchen: Kann als Risikofaktor für die Entwicklung von LHON beteiligt sein.
Übermäßiger Alkoholkonsum: Kann ebenfalls am Entwicklungsrisiko beteiligt sein.
Antituberkulose-Medikamente (Ethambutol usw.): Ein klinischer Zusammenhang wurde berichtet.
QWarum tritt die Harding-Krankheit häufiger bei Frauen auf?
A
LHON tritt überwiegend bei Männern auf (93,1 %), während bei der Harding-Krankheit 70,4 % der Patienten Frauen sind. Dies hängt damit zusammen, dass Multiple Sklerose (MS) häufiger bei Frauen vorkommt. Es wird vermutet, dass genetische und umweltbedingte Faktoren, die für MS prädisponieren, bei asymptomatischen Trägern von mtDNA-Mutationen LHON auslösen. 1) Auch eine Beteiligung der Östrogen-Signalübertragung an der Pathogenese der LHON wurde vorgeschlagen. 2)
Die Harding-Krankheit ist definiert als „ein Patient, der die MS-Diagnosekriterien erfüllt und eine wichtige LHON-Mutation aufweist“ (Pfeffer et al.).
In Japan gelten die 2015 etablierten Diagnosekriterien für LHON.
Bestätigter Fall : Erfüllt die Hauptkriterien (akute bis subakute, beidseitige, schmerzlose Sehverschlechterung mit Zentralskotom + mindestens ein ophthalmoskopischer Befund in der Akutphase).
Gesicherter Fall : Hauptsymptome + mitochondriale Gen-Missense-Mutation + MRT ohne retrobulbäre Sehnervenauffälligkeit
Verdachtsfall : mütterliche Vererbung offensichtlich, aber Genmutation nicht nachweisbar
Gentest: In Japan können Tests auf die drei Mutationen m.3460, m.11778 und m.14484 extern durchgeführt werden. Bei negativem Ergebnis ist eine Überweisung an ein Zentrum erforderlich.
Exom-Sequenzierung : Wenn keine mtDNA-Mutation identifiziert wird, werden Kern-Genmutationen wie DNAJC30 nachgewiesen.
MRT-Merkmale: Befunde, die die Harding-Krankheit von MS unterscheiden, umfassen eine verminderte Helligkeit und unscharfe Grenzen von T2-Läsionen, das Fehlen eines hohen Signals in T1-Läsionen und eine Zone hohen Signals um das Vorderhorn (anders als das typische Dawson-Finger-Muster). 1) 73 % bzw. 90 % der LHON-MS- und MS-Fälle erfüllen die McDonald-Kriterien für räumliche Dissemination. 1)
Kritische Flimmerfrequenz (CFF) und Pupillenlichtreflex-Test : erhalten oder leicht vermindert.
Screening-Überlegungen : Bei einem Screening von 1.666 MS-Patienten waren nur 5 LHON-Mutation-positiv, daher wird ein Routine-Screening nicht empfohlen. Eine Einzelfallentscheidung wird empfohlen, beschränkt auf Patienten mit schweren oder beidseitigen Gesichtsfeldausfällen oder mütterlicher Familienanamnese. 1)
Eine Differenzialdiagnose zur toxischen Optikusneuropathie, autosomal-dominanten Optikusatrophie (ADOA), okkulten Makuladystrophie, Zapfendystrophie, kompressiven Optikusneuropathie und Neuromyelitis-optica-Spektrum-Erkrankung (NMOSD) ist erforderlich.
Es gibt keine etablierte Behandlung. Nachfolgend sind die aktuellen Optionen aufgeführt.
Idebenon (nicht zugelassen) : Coenzym-Q10-Derivat. Es unterstützt den Elektronentransport und kann die Sehfunktion erhalten oder verbessern. In Japan wurden klinische Studien durchgeführt und in einigen Fällen wurde eine Verbesserung des Sehvermögens berichtet, aber das Medikament ist in Japan nicht zugelassen. Patienten können es persönlich importieren und einnehmen.
Nahrungsergänzungsmittel : Coenzym Q10, B-Vitamine, Vitamin C usw. werden nach Ermessen der jeweiligen Einrichtung verwendet.
Raucherentwöhnung: Da Rauchen das Risiko für LHON erhöht, wird eine Raucherentwöhnung empfohlen.
Genetische Beratung: Frühzeitig empfohlen für Frauen, die eine mtDNA-Mutation an ihre Nachkommen weitergeben könnten. Informieren, dass männliche Patienten die Mutation nicht an ihre Nachkommen vererben, dass es Fälle spontaner Heilung gibt und dass die Krankheit als seltene Krankheit anerkannt ist.
Low-Vision-Versorgung : Bereitstellung angemessener Betreuung und Lebensberatung für Patienten mit verbleibender Sehbehinderung.
Da keine Standardbehandlung etabliert ist, werden die folgenden Behandlungen versucht.
Methylprednisolon intravenös: Gabe von 1 g/Tag über 3 Tage, bei einigen Patienten wurde eine leichte bis mäßige Sehverbesserung berichtet. 1)
Mitoxantron : Etwa 19,2 mg/Monat intravenös zeigte eine Verbesserung des Sehvermögens und der neurologischen Symptome, aber aufgrund schwerwiegender Nebenwirkungen ist die Anwendung eingeschränkt. 1)
Plasmaaustausch und Cyclophosphamid : In einigen Fällen wurde eine subjektive Verbesserung der Lichtwahrnehmung und des Kontrastempfindens berichtet, jedoch nicht konsistent. 1)
Immunmodulatoren: Sie führen zu einer Stabilisierung der klinischen und MRT-Befunde, können jedoch das Fortschreiten der Sehbehinderung nicht aufhalten. Es gibt Berichte über einen Entzündungs-Rebound nach Absetzen von Natalizumab. 1)
QWelche Behandlung wird in Japan für die Harding-Krankheit durchgeführt?
A
Es gibt keine etablierte Behandlung; die Therapie ist hauptsächlich symptomatisch. Einige Patienten importieren persönlich das in Japan nicht zugelassene Idebenon zur oralen Einnahme, aber die Evidenz für die Wirksamkeit bei der Harding-Krankheit ist gering. 4)Derzeit werden Nahrungsergänzungsmittel wie CoQ10 und B-Vitamine, Raucherentwöhnung, Sehbehindertenversorgung und genetische Beratung angeboten.
Der grundlegende Mechanismus der LHON beruht auf einer mtDNA-Mutation, die zu einer Störung der Untereinheiten des Komplexes I (mitochondriale Atmungskette) führt. Der Defekt im Elektronentransport verringert die ATP-Synthese und führt gleichzeitig zur Akkumulation reaktiver Sauerstoffspezies (ROS). Dies induziert die Apoptose retinaler Ganglienzellen (RGC), was zur Optikusatrophie führt. 3)
Die Seherholungsraten variieren je nach Mutation: Die MT-ND4-Mutation hat eine Seherholungsrate von 4–25 %, niedriger als bei MT-ND1- und MT-ND6-Mutationen. 3)In Japan beträgt die Verbesserungsrate der Sehfunktion bei der häufigsten Mutation mt11778 (MT-ND4) nur wenige Prozent. Die Mutation mt14484 (MT-ND6) hat die höchste Erholungsrate.
Die höhere Penetranz der LHON bei Männern deutet auf eine Beteiligung von X-chromosomalen Kerngenen hin. 3)
PRICKLE3 (X-Chromosom, Xp11.23): reguliert die Funktion der ATP-Synthase (Komplex V). 3)
YARS2-Mutation : beeinträchtigt die Funktion der Komplexe I, III und IV der Elektronentransportkette. 3)
DNAJC30-Mutation (c.152A>G) : stört den Reparaturmechanismus von Komplex I und verursacht autosomal-rezessive LHON. 3)
Die Histopathologie der LHON-MS zeigt, dass T-Zellen und aktivierte Makrophagen/Mikroglia die Gewebeschädigung vermitteln. Das Vorhandensein von Entzündungszellen in LHON-Läsionen ist ungewöhnlich und deutet auf einen frühen immunologischen Mechanismus hin. 1) Die Veränderungen der weißen Substanz bestehen nicht nur aus MS-ähnlicher Demyelinisierung, sondern auch aus Vakuolisierung und diffuser Myelinblässe. Mitochondriale Dysfunktion, Autoimmunantwort und molekulare Mimikry werden als Mechanismen der entzündlichen Demyelinisierung vermutet. 1)
Die neurologischen Folgen reichen von leichten Beeinträchtigungen bis zu einem Verlauf ähnlich der schubförmig-remittierenden MS.
7. Aktuelle Forschung und Zukunftsperspektiven (Berichte aus der Forschungsphase)
Für LHON-Patienten mit MT-ND4-Mutation wurde eine Gentherapie (Lenadogen Nolparvovec; rAAV2/2-ND4) entwickelt. Sie führt durch allotope Genexpression das Wildtyp-ND4-Protein in die Mitochondrien der retinalen Ganglienzellen ein. 2)
Die REVERSE-Studie (2019) berichtete eine durchschnittliche BCVA-Verbesserung von -0,308 LogMAR in intravitreal injizierten Augen und -0,259 LogMAR in unbehandelten Augen. 2) Diese Ergebnisse deuten auf einen immunologischen Transfer zum kontralateralen Auge hin. Eine gepoolte Analyse (Studien RESCUE, REVERSE, RESTORE, REFLECT) zeigte eine Verbesserung von etwa 21,5 ETDRS-Buchstaben im Vergleich zur Kontrollgruppe. Die bilaterale Verabreichung war wirksamer als die unilaterale (ca. 12 vs. 8 ETDRS-Buchstaben), und eine Metaanalyse ergab, dass rAAV2/2-ND4 wirksamer als Idebenon war, beide wirksamer als der natürliche Verlauf. 2)
Derzeit nicht von EMA und FDA zugelassen, die Wirksamkeit bei Harding-Krankheit ist nicht verifiziert. Eine Wirkung auf die LHON-Komponente wird erwartet, während der Einfluss auf die MS-Pathologie unklar ist. 2)
Es wird eine Methode erforscht, bei der iPSC-abgeleitete MSC intravitreal verabreicht werden, um Mitochondrien direkt auf RGC zu übertragen. F-Aktin-abhängige tunneling nanotubes (TNT) vermitteln den Transfer. Bei Ndufs4-KO-Mäusen wurde eine Verhinderung der RGC-Dichteverringerung berichtet, jedoch wurde noch keine klinische Studie durchgeführt. 3)
Eine mögliche Verbesserung der visuell evozierten Potenziale (VEP) bei MS wurde angedeutet, und eine Anwendung bei Harding-Patienten mit schlechter visueller Prognose wird untersucht.
Die Entwicklung Harding-spezifischer Biomarker und prospektive Studien sind erforderlich. Die Behandlung dieser Erkrankung, die sowohl LHON als auch MS umfasst, sollte individualisiert werden, und eine multidisziplinäre Zusammenarbeit ist unerlässlich.
QKann die Gentherapie auch bei der Harding-Krankheit eingesetzt werden?
A
rAAV2/2-ND4 ist eine Gentherapie für LHON-Patienten mit MT-ND4-Mutation und wird eine Wirkung auf die LHON-Komponente erwartet. 2)Die Wirksamkeit bei der Harding-Krankheit ist jedoch nicht nachgewiesen, und sie ist derzeit von EMA und FDA nicht zugelassen. Der Einfluss auf die MS-Pathologie ist ebenfalls unklar, und eine sorgfältige Prüfung ist für die Anwendung bei dieser Erkrankung erforderlich.
Alorainy J, Alorfi Y, Karanjia R, Badeeb N. A Comprehensive Review of Leber Hereditary Optic Neuropathy and Its Association with Multiple Sclerosis-Like Phenotypes Known as Harding’s Disease. Eye and brain. 2024;16:17-24. doi:10.2147/EB.S470184. PMID:39100385; PMCID:PMC11296356.
Wong DCS, Makam R, Yu-Wai-Man P. Advanced therapies for inherited optic neuropathies. Eye (London, England). 2026;40(2):177-184. doi:10.1038/s41433-025-04109-1. PMID:41318849; PMCID:PMC12830909.
Hu JL, Hsu CC, Hsiao YJ, et al. Leber’s hereditary optic neuropathy: Update on the novel genes and therapeutic options. J Chin Med Assoc. 2024;87:12-16.
Sanders FWB, Votruba M. Outcomes of idebenone therapy for Leber hereditary optic neuropathy in a cohort of patients from Wales. Eye (Lond). 2025 Sep 17;39(16):2952-2957. doi:10.1038/s41433-025-03993-x. PMID:40962867; PMCID:PMC12583466.
Kopieren Sie den Artikeltext und fügen Sie ihn in den KI-Assistenten Ihrer Wahl ein.
Artikel in die Zwischenablage kopiert
Öffnen Sie unten einen KI-Assistenten und fügen Sie den kopierten Text in den Chat ein.