โรคฮาร์ดิงเป็นโรคหายากที่เกิดจากการกลายพันธุ์ของดีเอ็นเอไมโตคอนเดรีย (LHON ) ร่วมกับโรคปลอกประสาทเสื่อมแข็ง (MS )
มีรายงานผู้ป่วยในเอกสารเพียง 88 ราย โดย 70.4% เป็นเพศหญิง ซึ่งตรงกันข้ามกับ LHON ที่พบในเพศชายมากกว่า
อาการหลักคือการมองเห็น ลดลงแบบไม่เจ็บปวดในตาทั้งสองข้างและจุดบอดกลาง ตา โดยไม่มีอาการปวดตา แตกต่างจากโรคประสาทตาอักเสบ ใน MS
การกลายพันธุ์ที่พบบ่อยที่สุดคือ m.11778G>A (MT-ND4) ซึ่งพบใน 69.3% ของโรคฮาร์ดิง
ไม่มีวิธีการรักษาที่แน่ชัด ในประเทศญี่ปุ่นมีการใช้ไอเดเบโนน (ยังไม่ได้รับการรับรอง) อาหารเสริม CoQ10 การแนะนำให้เลิกสูบบุหรี่ และการดูแลผู้มีสายตาเลือนราง
เนื่องจากมีพยาธิสภาพทั้ง LHON และ MS ร่วมกัน จึงจำเป็นต้องได้รับการดูแลจากสหสาขาวิชาชีพ
แนะนำให้ให้คำปรึกษาทางพันธุกรรมแก่สตรีที่มีการกลายพันธุ์ตั้งแต่ระยะแรก
โรคฮาร์ดิง (Harding disease) เป็นโรคที่มีการกลายพันธุ์ของไมโตคอนเดรียดีเอ็นเอ (mtDNA) ในโรคจอประสาทตา เสื่อมทางพันธุกรรมแบบลีเบอร์ (Leber hereditary optic neuropathy; LHON ) ร่วมกับอาการทางระบบประสาทแบบทำลายปลอกไมอีลินที่คล้ายโรคปลอกประสาทเสื่อมแข็ง (multiple sclerosis; MS ) ตั้งชื่อตาม Harding และคณะที่รายงานในปี 1992 ถึงผู้หญิง 8 รายที่มีประวัติครอบครัวเป็น LHON และมีจอประสาทตา อักเสบทั้งสองข้าง (ในจำนวนนี้ 6 รายมีอาการทางระบบประสาทที่สอดคล้องกับ MS ) 1)
ในเอกสารทางการแพทย์มีรายงานผู้ป่วยทั้งหมด 88 ราย 1) ผู้ป่วย 70.4% (62 ราย) เป็นผู้หญิง อัตราส่วนเพศหญิงต่อชายคือ 2.38:1 1) ซึ่งตรงกันข้ามกับ LHON ที่พบในผู้ชายมากกว่า (ผู้ชาย 93.1%) และเชื่อว่าเกี่ยวข้องกับปัจจัยที่ทำให้ MS พบในผู้หญิงมากกว่า อายุเฉลี่ยที่เริ่มป่วยคือ 30.5 ปี 1)
ความชุกของ LHON ในญี่ปุ่นประมาณ 1/50,000 (คาดว่ามีผู้ป่วยทั้งหมดในประเทศประมาณ 4,000-5,000 คน) ในสหราชอาณาจักร 1/31,000 และในออสเตรเลีย 1/68,000 1) ในปี 2015 ญี่ปุ่นได้กำหนดให้ LHON เป็นโรคหายาก และได้กำหนดเกณฑ์การวินิจฉัยขึ้น จำนวนผู้ป่วยรายใหม่ประมาณ 117 คนต่อปี โดยร้อยละ 47 เกิดในผู้ที่มีอายุต่ำกว่า 30 ปี
Q
โรคฮาร์ดิงเป็นโรคที่พบได้น้อยเพียงใด?
A
มีรายงานผู้ป่วยในเอกสารเพียง 88 ราย ซึ่งเป็นโรคที่พบได้น้อยมาก 1) ผู้ป่วยร้อยละ 70.4 เป็นเพศหญิง อายุเฉลี่ยเมื่อเริ่มป่วยคือ 30.5 ปี เนื่องจากต้องมีการอยู่ร่วมกันของโรคสองชนิดคือ LHON และ MS จึงพบได้น้อยกว่า LHON หรือ MS เพียงอย่างเดียวมาก
Cristina Culea, Bogdana Tăbăcaru, Simona Stanca et al. Leber’s Hereditary
Optic Neuropathy – Case Discussion. Romanian Journal of Ophthalmology. 2019 Jan-Mar; 63(1):91. Figure 5. PMCID: PMC6531774. License: CC BY.
??????????????????????????โรคฮาร์ดิง?????????????????????
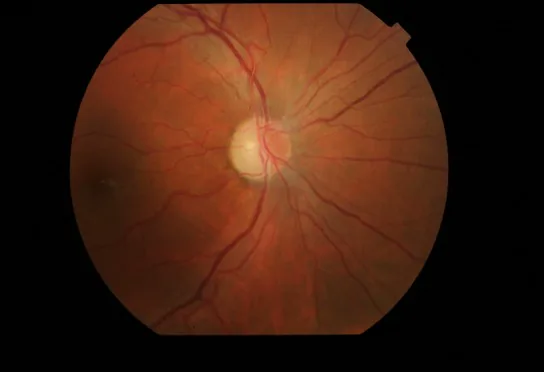
การมองเห็น ลดลงที่เริ่มจากภาพพร่ามัวการมองเห็น ลดลงอย่างรุนแรงและมีจุดบอดกลาง ภาพความผิดปกติของตาทั้งสองข้าง : มักส่งผลกระทบต่อตาทั้งสองข้าง ระยะเวลาก่อนที่ตาข้างที่สองจะได้รับผลกระทบจะนานกว่า LHON โดยเฉลี่ย 1.66 ปี 1) มีอาการผิดปกติทางการมองเห็น หลายครั้ง : ในขณะที่ LHON มักเกิดขึ้นเพียง 2 ครั้ง โรค Harding ทำให้เกิดอาการการมองเห็น ลดลงหลายครั้ง 1) ไม่มีอาการปวดตา : แตกต่างจากโรคประสาทตาอักเสบ ใน MS ตรงที่ไม่มีอาการปวดตา 1) นี่เป็นจุดสำคัญในการวินิจฉัยแยกโรคอาการนอกตา : อาจมีอาการสั่นขณะทรงท่า โรคปลายประสาทอักเสบ ความผิดปกติของการเคลื่อนไหว ภาวะหัวใจเต้นผิดจังหวะ กล้ามเนื้ออ่อนแรง และโรคกล้ามเนื้อร่วมด้วย
ระยะเฉียบพลัน
หัวประสาทตาแดง และคั่งเลือด : ร่วมกับอาการบวมของชั้นใยประสาทรอบหัวประสาทตา
การขยายตัวของหลอดเลือดฝอยจอตา : พบการขยายตัวของหลอดเลือดฝอยและการคดเคี้ยวของหลอดเลือดเพิ่มขึ้น
การไม่รั่วซึมในการตรวจฟลูออเรสซีน แองจิโอกราฟีของจอประสาทตา : ไม่พบการรั่วซึมของสีฟลูออเรสซีน จากหัวประสาทตาที่มีสีแดง ซึ่งเป็นจุดสำคัญในการแยกโรคนี้จากโรคประสาทตาอักเสบ
ผลการตรวจ OCT : พบว่ามีอาการบวมของหัวประสาทตาก่อนการเกิดโรค
ระยะเรื้อรัง
ฝ่อของหัวประสาทตา : หลังจากการอักเสบสงบลง จะเหลือรอยฝ่อของหัวประสาทตา
RNFL บางลงOCT พบว่าชั้นในของจอประสาทตา บางลง โดยเฉพาะบริเวณ papillo-macular bundle
การมองเห็น ที่ดีที่สุดหลังแก้ไขลดลง
การคงอยู่ของรีเฟล็กซ์รูม่านตา ต่อแสง : เมื่อเทียบกับโรคเส้นประสาทตา อื่นๆ รีเฟล็กซ์รูม่านตา ต่อแสงยังคงอยู่หรือมีความผิดปกติเพียงเล็กน้อย
ลานสายตา : แสดง central scotoma ที่หนาแน่น หรือ centrocecal scotoma
Q
การมองเห็นที่ลดลงในโรค Harding แตกต่างจาก LHON และโรคประสาทตาอักเสบใน MS อย่างไร?
A
ความแตกต่างจาก LHON คือ มีอาการมองเห็น ลดลงหลายครั้ง และระยะเวลาจนกระทั่งตาที่สองได้รับผลกระทบนั้นยาวนาน (เฉลี่ย 1.66 ปี) 1) ความแตกต่างจากโรคประสาทตาอักเสบ ใน MS คือ ไม่มีอาการปวดตา และไม่พบการรั่วของสีจากหัวประสาทตาในการตรวจฟลูออเรสซีน แองจิโอกราฟี ลักษณะเหล่านี้เป็นเบาะแสสำคัญในการแยกโรค Harding
สาเหตุของโรค Harding คือการกลายพันธุ์ที่ก่อโรคในไมโตคอนเดรีย DNA (mtDNA) การกลายพันธุ์หลักสามชนิดล้วนเข้ารหัสซับยูนิตของคอมเพล็กซ์ I ทำให้เกิดการสังเคราะห์ ATP บกพร่องและเพิ่มอนุมูลอิสระ (ROS) ซึ่งนำไปสู่การตายแบบอะพอพโทซิส ของเซลล์ปมประสาทจอตา (RGC ) 3)
สัดส่วนของการกลายพันธุ์ในผู้ป่วยโรค Harding 88 รายแสดงดังนี้
การกลายพันธุ์ ยีน สัดส่วนในโรค Harding m.11778G>A MT-ND4 69.3% (61/88 ราย) m.14484T>C MT-ND6 12.5% m.3460G>A MT-ND1 10.2%
อ้างอิง: Alorainy J et al. 20241)
การกลายพันธุ์ MT-ND4 คิดเป็น 90% ของผู้ป่วย LHON ทั้งหมดในเอเชีย และ 70% ในยุโรป 3) ในผู้ป่วยในประเทศ การกลายพันธุ์ของยีนทั้งสามนี้คิดเป็น 95%
mtDNA ถ่ายทอดจากแม่สู่ลูก (การถ่ายทอดทางมารดา) ไม่ถ่ายทอดไปยังลูกหลานของผู้ป่วยชาย
กลไกการอยู่ร่วมกันของ LHON และ MS มีสมมติฐานสามข้อดังต่อไปนี้ 1)
ทฤษฎีการปรับเปลี่ยน LHON โดย MS : การกลายพันธุ์ของ mtDNA ปรับเปลี่ยนฟีโนไทป์ของ MS ทำให้เกิดโรคประสาทตาอักเสบ ผิดปกติที่ไม่เจ็บปวด รุนแรงกว่า และไม่สามารถกลับคืนได้ทฤษฎีการแสดงออกของ LHON เนื่องจากปัจจัยโน้มเอียงของ MS : ปัจจัยทางพันธุกรรมและสิ่งแวดล้อมที่ทำให้เกิดปัจจัยโน้มเอียงของ MS ในผู้หญิง ส่งเสริมการเกิด LHON ในผู้ที่มีการกลายพันธุ์ของ mtDNA ที่ไม่มีอาการทฤษฎีการเหนี่ยวนำการอักเสบโดยบังเอิญ : ในผู้ป่วยที่มีการกลายพันธุ์ของ mtDNA ของ LHON จะเกิดปฏิกิริยาการอักเสบโดยบังเอิญในทางเดินประสาทตาส่วนหน้า
การสูบบุหรี่ : อาจเกี่ยวข้องกับความเสี่ยงในการเกิด LHON การดื่มแอลกอฮอล์ในปริมาณมาก : อาจเกี่ยวข้องกับความเสี่ยงในการเกิดโรคเช่นกันยาต้านวัณโรค (เช่น Ethambutol) : มีรายงานว่ามีความเกี่ยวข้องทางคลินิก
ผู้ที่มียีน mtDNA กลายพันธุ์ควรหลีกเลี่ยงการสูบบุหรี่และการดื่มแอลกอฮอล์ในปริมาณมาก ควรตรวจตาเป็นประจำก่อนเริ่มมีอาการเพื่อสังเกตการเปลี่ยนแปลงของการมองเห็น ได้เร็ว
Q
ทำไมโรค Harding จึงพบในผู้หญิงมากกว่า?
A
LHON พบในผู้ชายมากกว่า (ผู้ชาย 93.1%) แต่โรค Harding พบในผู้หญิง 70.4% ซึ่งสัมพันธ์กับโรค MS ที่พบในผู้หญิงมากกว่า มีสมมติฐานว่าปัจจัยทางพันธุกรรมและสิ่งแวดล้อมที่ทำให้เกิด MS อาจกระตุ้นให้เกิด LHON ในผู้ที่มียีน mtDNA กลายพันธุ์โดยไม่มีอาการ 1) นอกจากนี้ยังมีข้อเสนอว่าการส่งสัญญาณของเอสโตรเจนอาจเกี่ยวข้องกับพยาธิสภาพของ LHON 2)
โรค Harding ถูกกำหนดให้เป็น “ผู้ป่วยที่เข้าเกณฑ์การวินิจฉัย MS และมีการกลายพันธุ์ LHON หลัก” (Pfeffer และคณะ)
ในญี่ปุ่น ใช้เกณฑ์การวินิจฉัย LHON ที่กำหนดขึ้นในปี 2015
กรณียืนยัน : ผู้ที่มีอาการหลัก (การมองเห็น ลดลงแบบเฉียบพลันถึงกึ่งเฉียบพลัน ตาทั้งสองข้าง ไม่เจ็บปวด และมีจุดบอดกลาง ร่วมกับความผิดปกติจากการตรวจตาในระยะเฉียบพลันอย่างน้อย 1 ข้อ)กรณีที่แน่นอน : อาการหลัก + การกลายพันธุ์แบบมิสเซนส์ของยีนไมโตคอนเดรีย + MRI ไม่พบความผิดปกติของเส้นประสาทตา ส่วนหลังลูกตากรณีที่สงสัย : มีการถ่ายทอดทางมารดาชัดเจน แต่ตรวจไม่พบการกลายพันธุ์ของยีน
การตรวจ วัตถุประสงค์/ผลการตรวจที่สำคัญ การวิเคราะห์ mtDNA และแผงยีนหลายตัว การระบุ m.3460G>A, m.11778G>A, m.14484T>C MRI การตรวจหารอยโรคเนื้อขาวสัญญาณสูง T2 แบบทำลายไมอีลิน การตรวจหลอดเลือดด้วยฟลูออเรสซีน การรั่วของสีจากหัวประสาทตา (การไม่รั่วเป็นจุดแยก) เครื่องตรวจการเชื่อมโยงแสง (OCT ) การบางลงของมัดใยประสาทจากจอประสาทตา ถึงหัวประสาทตา คลื่นไฟฟ้าหัวใจ (ECG) การแยกโรคหัวใจเต้นเร็วผิดปกติจากกลุ่มอาการก่อนการกระตุ้นหัวใจ
การตรวจทางพันธุกรรม : ในประเทศญี่ปุ่นสามารถส่งตรวจการกลายพันธุ์ 3 ชนิด ได้แก่ m.3460, m.11778, m.14484 จากภายนอกได้ หากผลเป็นลบ จำเป็นต้องส่งต่อสถานพยาบาลหลักการหาลำดับเอ็กโซม : หากไม่พบการกลายพันธุ์ของ mtDNA ให้ตรวจหายีนในนิวเคลียส เช่น DNAJC30ลักษณะ MRI : สิ่งที่ช่วยแยกโรค Harding จาก MS ได้แก่ ความเข้มของรอยโรค T2 ลดลงและขอบเขตไม่ชัดเจน การไม่มีสัญญาณความเข้มสูงในรอยโรค T1 และบริเวณสัญญาณความเข้มสูงรอบ anterior horn (แตกต่างจากรูปแบบ Dawson’s fingers ทั่วไป) 1) LHON -MS และ MS ร้อยละ 73 และ 90 ตามลำดับเข้าเกณฑ์การแพร่กระจายเชิงพื้นที่ของ McDonald 1) ค่า CFF และการตรวจ pupillary light reflex : ยังคงปกติหรือลดลงเล็กน้อยการพิจารณาคัดกรอง : จากการคัดกรองผู้ป่วย MS 1,666 ราย พบการกลายพันธุ์ LHON เพียง 5 ราย ดังนั้นจึงไม่แนะนำให้คัดกรองเป็นประจำ แนะนำให้พิจารณาเป็นรายกรณีเฉพาะในผู้ป่วยที่มีความบกพร่องลานสายตารุนแรงหรือเป็นสองตา หรือมีประวัติครอบครัวทางมารดา 1)
จำเป็นต้องแยกจากภาวะเส้นประสาทตา อักเสบจากพิษ ภาวะจอประสาทตา เสื่อมชนิดถ่ายทอดทางพันธุกรรมแบบเด่น (ADOA) ภาวะจอประสาทตา เสื่อมชนิดออคคัลท์ ภาวะจอประสาทตา เสื่อมชนิดรูปกรวย ภาวะเส้นประสาทตา ถูกกดทับ และกลุ่มอาการอักเสบของเส้นประสาทตา และไขสันหลัง (NMOSD )
ยังไม่มีการรักษาที่เป็นมาตรฐานที่แน่ชัด ต่อไปนี้เป็นแนวทางการจัดการในปัจจุบัน
ไอดีเบโนน (ยังไม่ได้รับการอนุมัติ) : อนุพันธ์ของโคเอ็นไซม์ Q10 ช่วยในการถ่ายโอนอิเล็กตรอน และอาจช่วยรักษาหรือปรับปรุงการมองเห็น มีการทดลองทางคลินิกในญี่ปุ่นและรายงานการปรับปรุงการมองเห็น ในบางกรณี แต่เป็นยาที่ยังไม่ได้รับการอนุมัติในประเทศ ผู้ป่วยอาจนำเข้าด้วยตนเองเพื่อรับประทานอาหารเสริม : โคเอ็นไซม์ Q10 วิตามินบีรวม และวิตามินซี ใช้ตามดุลยพินิจของแต่ละสถานพยาบาลคำแนะนำให้เลิกสูบบุหรี่ : เนื่องจากการสูบบุหรี่เกี่ยวข้องกับความเสี่ยงในการเกิด LHON จึงแนะนำให้เลิกสูบบุหรี่การให้คำปรึกษาทางพันธุกรรม การดูแลผู้มีความบกพร่องทางการมองเห็น : ให้การดูแลที่เหมาะสมและคำแนะนำในการใช้ชีวิตแก่ผู้ป่วยที่ยังมีความบกพร่องทางการมองเห็น หลงเหลืออยู่
ในขณะที่ยังไม่มีการรักษามาตรฐานที่ชัดเจน ได้มีการทดลองรักษาด้วยวิธีดังต่อไปนี้
การให้เมทิลเพรดนิโซโลนทางหลอดเลือดดำ : การให้ขนาด 1 กรัม/วัน เป็นเวลา 3 วัน มีรายงานว่าช่วยให้การมองเห็น ดีขึ้นเล็กน้อยถึงปานกลางในผู้ป่วยบางราย 1) มิโทแซนโทรน : มีรายงานการฟื้นฟูการมองเห็น และการปรับปรุงอาการทางระบบประสาทด้วยการฉีดเข้าหลอดเลือดดำประมาณ 19.2 มก./เดือน แต่การใช้มีจำกัดเนื่องจากผลข้างเคียงที่รุนแรง 1) การแลกเปลี่ยนพลาสมา /ไซโคลฟอสฟาไมด์1) ยากดภูมิคุ้มกัน : ทำให้อาการทางคลินิกและผล MRI คงที่ แต่ไม่สามารถยับยั้งการลุกลามของความบกพร่องทางการมองเห็น มีรายงานการอักเสบกลับมาหลังหยุดนาทาลิซูแมบ 1)
Q
ในญี่ปุ่นมีการรักษาโรค Harding อย่างไร?
A
ยังไม่มีวิธีการรักษาที่เป็นมาตรฐาน การรักษาส่วนใหญ่เป็นการรักษาตามอาการ ผู้ป่วยบางรายนำเข้าไอเดเบโนนที่ยังไม่ได้รับการอนุมัติในประเทศมารับประทานเอง แต่หลักฐานประสิทธิภาพในโรค Harding มีน้อย 4) ปัจจุบันมีการให้อาหารเสริม เช่น CoQ10 และวิตามินบีรวม การแนะนำให้เลิกบุหรี่ การดูแลสายตาเลือนราง และการให้คำปรึกษาทางพันธุกรรม
กลไกพื้นฐานของ LHON เริ่มต้นจากความผิดปกติของหน่วยย่อยของคอมเพล็กซ์ I (สายโซ่การหายใจของไมโตคอนเดรีย) เนื่องจากการกลายพันธุ์ของ mtDNA การรบกวนการถ่ายเทอิเล็กตรอนทำให้การสังเคราะห์ ATP ลดลง และในขณะเดียวกันก็เกิดการสะสมของอนุมูลอิสระ (ROS) ซึ่งกระตุ้นให้ RGC เกิดอะพอพโทซิส นำไปสู่การฝ่อของเส้นประสาทตา 3)
อัตราการฟื้นตัวของการมองเห็น แตกต่างกันไปตามชนิดของการกลายพันธุ์ โดยการกลายพันธุ์ MT-ND4 มีอัตราการฟื้นตัว 4-25% ซึ่งต่ำกว่าการกลายพันธุ์ MT-ND1 และ MT-ND6 3) ในประเทศญี่ปุ่น การกลายพันธุ์ mt11778 (MT-ND4) ที่พบบ่อยที่สุดมีอัตราการฟื้นตัวของการมองเห็น เพียงไม่กี่เปอร์เซ็นต์ การกลายพันธุ์ mt14484 (MT-ND6) มีอัตราการฟื้นตัวสูงที่สุด
เนื่องจากอัตราการแทรกซึมของ LHON สูงในเพศชาย จึงมีการเสนอว่ายีนนิวเคลียร์ที่เชื่อมโยงกับโครโมโซม X มีส่วนเกี่ยวข้อง 3)
PRICKLE3 (โครโมโซม X ตำแหน่ง Xp11.23): ควบคุมการทำงานของเอนไซม์ ATP synthase (คอมเพล็กซ์ V) 3) การกลายพันธุ์ YARS2 : ทำให้การทำงานของคอมเพล็กซ์ I, III, IV ในระบบขนส่งอิเล็กตรอนบกพร่อง 3) การกลายพันธุ์ DNAJC30 (c.152A>G): ทำให้กลไกซ่อมแซมคอมเพล็กซ์ I บกพร่อง และทำให้เกิด LHON แบบถ่ายทอดทาง autosomal recessive 3)
พยาธิวิทยาของเนื้อเยื่อใน LHON -MS พบว่า T cells และ activated macrophages/microglia เป็นสื่อกลางในการทำลายเนื้อเยื่อ การมีเซลล์อักเสบภายในรอยโรค LHON เป็นสิ่งผิดปกติ บ่งชี้ถึงกลไกทางภูมิคุ้มกันในระยะเริ่มต้น 1) การเปลี่ยนแปลงของสารสีขาวไม่เพียงแต่เกิดจากการทำลายปลอกไมอีลินแบบ MS แต่ยังเกิดจาก vacuolation และ diffuse myelin pallor ร่วมด้วย ความผิดปกติของไมโตคอนเดรีย การตอบสนองของภูมิคุ้มกันตนเอง และ molecular mimicry เป็นกลไกที่สันนิษฐานของการทำลายปลอกไมอีลินแบบอักเสบ 1)
ผลกระทบทางระบบประสาทที่เหลืออยู่มีตั้งแต่ความบกพร่องเล็กน้อยไปจนถึงอาการคล้ายโรค MS แบบ relapsing-remitting
เนื้อหาต่อไปนี้อยู่ในระยะวิจัยหรือการทดลองทางคลินิกเท่านั้น และไม่ใช่การรักษามาตรฐานที่สามารถรับได้ในโรงพยาบาลทั่วไป เป็นข้อมูลอ้างอิงสำหรับผู้เชี่ยวชาญเกี่ยวกับการพัฒนาทางการแพทย์ในอนาคต
การรักษาด้วยยีนบำบัด (เลนาโดจีน นอร์พาร์โวเวก; rAAV2/2-ND4) ได้รับการพัฒนาสำหรับผู้ป่วย LHON ที่มีการกลายพันธุ์ของ MT-ND4 โดยใช้การแสดงออกของยีนแบบอัลโลโทปิกเพื่อนำโปรตีน ND4 ชนิดปกติเข้าสู่ไมโทคอนเดรียของ RGC 2)
การศึกษา REVERSE (2019) รายงานว่าค่าเฉลี่ย BCVA ที่ดีขึ้นในตาที่ได้รับการฉีดเข้าแก้วตาเท่ากับ -0.308 LogMAR และในตาที่ไม่ได้รับการรักษาเท่ากับ -0.259 LogMAR 2) ผลลัพธ์นี้บ่งชี้ถึงการถ่ายโอนทางภูมิคุ้มกันไปยังตาอีกข้างหนึ่ง การวิเคราะห์แบบรวม (จากการศึกษา RESCUE, REVERSE, RESTORE และ REFLECT จำนวน 4 การศึกษา) แสดงให้เห็นว่ากลุ่มที่ได้รับการรักษามีการปรับปรุงดีขึ้นประมาณ 21.5 ตัวอักษร ETDRS เมื่อเทียบกับกลุ่มควบคุม การให้ยาทั้งสองข้างมีประสิทธิภาพมากกว่าการให้ยาข้างเดียว (ประมาณ 12 เทียบกับประมาณ 8 ตัวอักษร ETDRS) และการวิเคราะห์เมตาก็พบว่า rAAV2/2-ND4 มีประสิทธิภาพมากกว่า idebenone และทั้งสองวิธีมีประสิทธิภาพมากกว่าการดำเนินโรคตามธรรมชาติ 2)
ปัจจุบันยังไม่ได้รับการอนุมัติจาก EMA และ FDA และยังไม่มีการตรวจสอบประสิทธิภาพสำหรับโรค Harding คาดว่าจะมีผลต่อส่วนประกอบของ LHON แต่ผลต่อพยาธิสภาพของ MS ยังไม่ชัดเจน 2)
กำลังศึกษาวิธีการฉีด MSC ที่ได้จาก iPSC เข้าไปในวุ้นตา เพื่อถ่ายเทไมโทคอนเดรียโดยตรงไปยัง RGC โดยท่อนาโนแบบอุโมงค์ที่ขึ้นกับ F-actin (TNT) เป็นตัวกลางในการถ่ายเท ในหนู Ndufs4 KO มีรายงานว่าสามารถป้องกันการลดลงของความหนาแน่น RGC แต่ยังไม่ถึงขั้นทดลองทางคลินิก 3)
มีการเสนอแนะถึงความเป็นไปได้ในการปรับปรุงศักยภาพที่เกิดจากการมองเห็น (VEP ) ใน MS และกำลังพิจารณาการประยุกต์ใช้ในผู้ป่วยโรค Harding ที่มีการพยากรณ์โรคทางสายตาไม่ดี
จำเป็นต้องมีการพัฒนาไบโอมาร์กเกอร์เฉพาะสำหรับโรค Harding และการศึกษาวิจัยแบบไปข้างหน้า การรักษาสำหรับโรคนี้ซึ่งมีพยาธิสภาพสองอย่างคือ LHON และ MS ควรเป็นแบบเฉพาะบุคคล และการทำงานร่วมกันของสหสาขาวิชาชีพเป็นสิ่งที่ขาดไม่ได้
Q
การบำบัดด้วยยีนสามารถใช้กับโรค Harding ได้หรือไม่?
A
rAAV2/2-ND4 เป็นการบำบัดด้วยยีน สำหรับผู้ป่วย LHON ที่มีการกลายพันธุ์ของ MT-ND4 ซึ่งคาดว่าจะมีผลต่อองค์ประกอบของ LHON 2) อย่างไรก็ตาม ยังไม่มีการตรวจสอบประสิทธิภาพในโรค Harding และยังไม่ได้รับการอนุมัติจาก EMA และ FDA ในปัจจุบัน ผลกระทบต่อพยาธิสภาพของ MS ก็ยังไม่ชัดเจน และจำเป็นต้องพิจารณาอย่างรอบคอบในการประยุกต์ใช้กับโรคนี้
Alorainy J, Alorfi Y, Karanjia R, Badeeb N. A Comprehensive Review of Leber Hereditary Optic Neuropathy and Its Association with Multiple Sclerosis-Like Phenotypes Known as Harding’s Disease. Eye and brain. 2024;16:17-24. doi:10.2147/EB .S470184. PMID:39100385; PMCID:PMC11296356.
Wong DCS, Makam R, Yu-Wai-Man P. Advanced therapies for inherited optic neuropathies. Eye (London, England). 2026;40(2):177-184. doi:10.1038/s41433-025-04109-1. PMID:41318849; PMCID:PMC12830909.
Hu JL, Hsu CC, Hsiao YJ, et al. Leber’s hereditary optic neuropathy: Update on the novel genes and therapeutic options. J Chin Med Assoc. 2024;87:12-16.
Sanders FWB, Votruba M. Outcomes of idebenone therapy for Leber hereditary optic neuropathy in a cohort of patients from Wales. Eye (Lond). 2025 Sep 17;39(16):2952-2957. doi:10.1038/s41433-025-03993-x. PMID:40962867; PMCID:PMC12583466.
ถาม AI เกี่ยวกับบทความนี้
คัดลอกข้อความบทความแล้ววางในผู้ช่วย AI ที่คุณต้องการใช้
เปิดผู้ช่วย AI ด้านล่าง แล้ววางข้อความที่คัดลอกลงในช่องแชต