La maladie de Harding est une affection dans laquelle des symptômes neurologiques démyélinisants similaires à la sclérose en plaques (SEP) coexistent avec une mutation de l’ADN mitochondrial (ADNmt) de la neuropathie optique héréditaire de Leber (LHON). Elle tire son nom du rapport de Harding et al. en 1992, qui ont décrit huit femmes atteintes de neuropathie optique bilatérale avec des antécédents familiaux de LHON (dont six présentaient des symptômes neurologiques compatibles avec la SEP). 1)
Au total, 88 cas ont été rapportés dans la littérature. 1) 70,4 % (62 cas) des patients sont des femmes, avec un sex-ratio de 2,38:1. 1) Cela contraste avec la LHON, qui touche principalement les hommes (93,1 %), et suggère l’implication d’une prédisposition féminine à la SEP. L’âge moyen d’apparition est de 30,5 ans. 1)
La prévalence de la LHON est estimée à environ 1/50 000 au Japon (soit environ 4 000 à 5 000 patients au total), 1/31 000 au Royaume-Uni et 1/68 000 en Australie. 1) En 2015, la LHON a été désignée maladie rare au Japon et des critères diagnostiques ont été établis. Le nombre de nouveaux cas est d’environ 117 par an, dont 47 % surviennent avant l’âge de 30 ans.
QQuelle est la rareté de la maladie de Harding ?
A
Seulement 88 cas ont été rapportés dans la littérature, ce qui en fait une maladie extrêmement rare. 1) 70,4 % des patients sont des femmes et l’âge moyen d’apparition est de 30,5 ans. Comme elle nécessite la coexistence de la LHON et de la SEP, elle est beaucoup moins fréquente que la LHON ou la SEP seules.
Baisse de l’acuité visuelle débutant par un brouillard visuel : commence par un brouillard visuel indolore, s’aggrave progressivement pour aboutir à une baisse sévère de l’acuité visuelle et un scotome central.
Atteinte bilatérale : affecte généralement les deux yeux. L’intervalle avant l’atteinte du deuxième œil est plus long que dans la LHON, avec une moyenne de 1,66 an. 1)
Épisodes multiples de troubles visuels : alors que la LHON n’en provoque généralement que deux, la maladie de Harding entraîne plusieurs épisodes de baisse de l’acuité visuelle. 1)
Sans douleur oculaire : Contrairement à la névrite optique de la SEP, elle n’est pas accompagnée de douleur oculaire. 1) C’est un point clé pour le diagnostic différentiel.
Symptômes extra-oculaires : Peut être accompagné de tremblements posturaux, neuropathie périphérique, troubles moteurs, arythmie cardiaque, faiblesse musculaire, maladies musculaires.
Signes cliniques (observés par le médecin lors de l’examen)
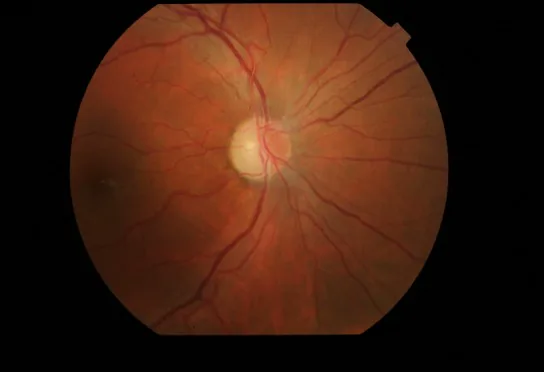
Rougeur et hyperhémie de la papille optique : Avec œdème de la couche des fibres nerveuses rétiniennes (RNFL) péripapillaire.
Télangiectasies rétiniennes : Dilatation des capillaires (télangiectasie) et augmentation de la tortuosité vasculaire.
Absence de fuite à l’angiographie à la fluorescéine : Aucune fuite de colorant fluorescent n’est observée à partir de la papille optique hyperhémiée. C’est un point de différenciation important avec la névrite optique.
Résultats OCT : Un œdème de la papille optique serait observé avant l’apparition des symptômes.
Phase chronique
Atrophie de la papille optique : Une atrophie papillaire persiste après la résolution de l’inflammation.
Amincissement de la RNFL : L’OCT montre un amincissement des couches internes de la rétine, principalement au niveau du faisceau papillo-maculaire.
Baisse de l’acuité visuelle corrigée : Elle reste souvent autour de 0,01. La perception lumineuse est préservée.
Préservation du réflexe pupillaire à la lumière : par rapport aux autres maladies du nerf optique, le réflexe pupillaire à la lumière est conservé ou seulement légèrement altéré.
Champ visuel : présente un scotome central dense ou un scotome centrocaecal (centrocecal scotoma).
QEn quoi la baisse de vision dans la maladie de Harding diffère-t-elle de celle de la LHON ou de la névrite optique de la SEP ?
A
La différence avec la LHON réside dans la survenue de multiples épisodes de baisse de vision et un intervalle plus long avant l’atteinte du deuxième œil (moyenne 1,66 an). 1) La différence avec la névrite optique de la SEP est l’absence de douleur oculaire et l’absence de fuite de colorant de la papille optique à l’angiographie à la fluorescéine. Ces caractéristiques sont des indices importants pour le diagnostic différentiel de la maladie de Harding.
La maladie de Harding est causée par des mutations pathogènes de l’ADN mitochondrial (ADNmt). Les trois mutations principales codent toutes pour des sous-unités du complexe I, induisant une apoptose des cellules ganglionnaires rétiniennes (CGR) via un déficit de synthèse d’ATP et une augmentation des espèces réactives de l’oxygène (ROS). 3)
La répartition des mutations dans 88 cas de maladie de Harding est présentée ci-dessous.
Mutation
Gène
Proportion dans la maladie de Harding
m.11778G>A
MT-ND4
69,3 % (61/88 cas)
m.14484T>C
MT-ND6
12,5 %
m.3460G>A
MT-ND1
10,2 %
Citation : Alorainy J et al. 20241)
La mutation MT-ND4 représente 90 % des cas de LHON en Asie et 70 % en Europe.3) Chez les patients japonais, ces trois mutations génétiques représentent également 95 % des cas.
L’ADNmt est transmis de la mère à l’enfant (hérédité maternelle). Il n’est pas transmis à la descendance des patients masculins.
Hypothèse pathologique de la coexistence de LHON et de SEP
Trois hypothèses ont été proposées pour expliquer le mécanisme de coexistence de LHON et de SEP.1)
Hypothèse de modification de LHON par la SEP : la mutation de l’ADNmt modifie le phénotype de la SEP, provoquant une névrite optique atypique, indolore, plus sévère et irréversible.
Théorie de la manifestation de la LHON due à une prédisposition à la SEP : Chez les femmes, les facteurs génétiques et environnementaux prédisposant à la SEP favorisent l’apparition de la LHON chez les porteurs asymptomatiques de mutations de l’ADNmt.
Théorie de l’induction inflammatoire accidentelle : Chez les patients porteurs de mutations de l’ADNmt de la LHON, une réaction inflammatoire accidentelle est induite dans les voies visuelles antérieures.
Tabagisme : Peut être impliqué comme facteur de risque de développement de la LHON.
Consommation excessive d’alcool : Peut également être impliquée dans le risque de développement.
Médicaments antituberculeux (éthambutol, etc.) : Une implication clinique a été signalée.
QPourquoi la maladie de Harding est-elle plus fréquente chez les femmes ?
A
La LHON est prédominante chez les hommes (93,1 %), mais dans la maladie de Harding, 70,4 % des patients sont des femmes. Cela est lié au fait que la sclérose en plaques (SEP) est plus fréquente chez les femmes. Une hypothèse suggère que des facteurs génétiques et environnementaux prédisposant à la SEP déclenchent la LHON chez les porteurs asymptomatiques de mutations de l’ADNmt. 1) Il a également été suggéré que la signalisation des œstrogènes pourrait être impliquée dans la pathogenèse de la LHON. 2)
La maladie de Harding est définie comme « un patient répondant aux critères diagnostiques de la SEP et présentant une mutation LHON majeure » (Pfeffer et al.).
Au Japon, les critères diagnostiques de la LHON établis en 2015 sont appliqués.
Cas confirmé : répond aux critères principaux (baisse de l’acuité visuelle aiguë à subaiguë, bilatérale, indolore avec scotome central + au moins une anomalie ophtalmoscopique en phase aiguë).
Cas certain : signes principaux + mutation faux-sens du gène mitochondrial + IRM sans anomalie du nerf optique rétrobulbaire
Cas suspect : hérédité maternelle évidente mais mutation génétique non détectable
Test génétique : au Japon, les tests pour les trois mutations m.3460, m.11778 et m.14484 peuvent être sous-traités. En cas de résultat négatif, une demande auprès d’un établissement de référence est nécessaire.
Séquençage de l’exome : si une mutation de l’ADNmt n’est pas identifiée, détecter des mutations de gènes nucléaires comme DNAJC30.
Caractéristiques IRM : Les signes permettant de distinguer la maladie de Harding de la SEP comprennent une diminution de l’intensité des lésions T2 et des limites floues, l’absence de signal élevé dans les lésions T1, et une zone de signal élevé autour de la corne antérieure (différente du motif typique des doigts de Dawson). 1) 73 % et 90 % des cas de LHON-SEP et de SEP répondent aux critères de dissémination spatiale de McDonald. 1)
Valeur de fréquence critique de fusion (CFF) et test de réflexe pupillaire : conservée ou légèrement diminuée.
Dépistage : Sur 1 666 patients atteints de SEP, seuls 5 présentaient une mutation LHON positive, ce qui rend le dépistage systématique non recommandé. Une décision au cas par cas est recommandée, limitée aux patients présentant un déficit du champ visuel sévère ou bilatéral, ou des antécédents familiaux maternels. 1)
Il n’existe pas de traitement établi. Voici les options actuelles.
Idébénone (non approuvé) : Dérivé de la coenzyme Q10. Il aide le transport des électrons et peut maintenir ou améliorer la fonction visuelle. Des essais cliniques ont été menés au Japon et certaines améliorations visuelles ont été rapportées, mais ce médicament n’est pas approuvé au Japon. Certains patients l’importent pour usage personnel.
Compléments alimentaires : Coenzyme Q10, vitamines B, vitamine C, etc., sont utilisés selon le jugement de chaque établissement.
Conseil d’arrêt du tabac : Le tabagisme étant un facteur de risque de développer la LHON, il est conseillé d’arrêter de fumer.
Conseil génétique : recommandé précocement aux femmes susceptibles de transmettre une mutation de l’ADNmt à leur descendance. Informer que les hommes ne transmettent pas la mutation à leurs enfants, qu’il existe des cas de guérison spontanée et que la maladie est reconnue comme maladie rare.
Soins de basse vision : fournir des soins appropriés et des conseils de vie aux patients présentant une déficience visuelle résiduelle.
En l’absence de traitement standard établi, les traitements suivants sont essayés.
Méthylprednisolone intraveineuse : administration de 1 g/jour pendant 3 jours, une amélioration légère à modérée de l’acuité visuelle a été rapportée chez certains patients. 1)
Mitoxantrone : environ 19,2 mg/mois par voie intraveineuse a montré une amélioration de l’acuité visuelle et des symptômes neurologiques, mais son utilisation est limitée en raison d’effets secondaires graves. 1)
Échange plasmatique et cyclophosphamide : certaines améliorations subjectives de la perception lumineuse et de la sensibilité au contraste ont été rapportées, mais elles ne sont pas cohérentes. 1)
Immunomodulateurs : apportent une stabilité clinique et IRM, mais ne peuvent pas empêcher la progression des troubles visuels. Des cas de rebond inflammatoire après l’arrêt du natalizumab ont été rapportés. 1)
QQuels traitements sont pratiqués au Japon pour la maladie de Harding ?
A
Il n’existe pas de traitement standardisé ; les soins sont principalement symptomatiques. Certains patients importent personnellement de l’idébénone, non approuvé au Japon, pour le prendre par voie orale, mais les preuves d’efficacité dans la maladie de Harding sont limitées. 4)Actuellement, des suppléments comme le CoQ10 et les vitamines B, des conseils pour arrêter de fumer, des soins de basse vision et un conseil génétique sont proposés.
Le mécanisme de base de la LHON provient d’une mutation de l’ADNmt entraînant un dysfonctionnement des sous-unités du complexe I (chaîne respiratoire mitochondriale). Le défaut de transport d’électrons réduit la synthèse d’ATP et accumule simultanément des espèces réactives de l’oxygène (ROS). Cela induit l’apoptose des cellules ganglionnaires rétiniennes (CGR), conduisant à une atrophie du nerf optique. 3)
Les taux de récupération visuelle varient selon la mutation : la mutation MT-ND4 présente un taux de récupération de 4 à 25 %, inférieur à celui des mutations MT-ND1 et MT-ND6. 3)Au Japon, la mutation la plus fréquente, mt11778 (MT-ND4), n’a qu’un taux d’amélioration visuelle de quelques pour cent. La mutation mt14484 (MT-ND6) a le taux de récupération le plus élevé.
La pénétrance plus élevée de la LHON chez les hommes suggère l’implication de gènes nucléaires liés au chromosome X. 3)
PRICKLE3 (chromosome X, Xp11.23) : régule la fonction de l’ATP synthase (complexe V). 3)
Mutation YARS2 : altère les fonctions des complexes I, III et IV de la chaîne respiratoire. 3)
Mutation DNAJC30 (c.152A>G) : perturbe le mécanisme de réparation du complexe I, provoquant une LHON autosomique récessive. 3)
L’histopathologie de la LHON-MS montre que les lymphocytes T et les macrophages/microglies activés sont les médiateurs des lésions tissulaires. La présence de cellules inflammatoires dans les lésions LHON est inhabituelle et suggère un mécanisme immunologique précoce. 1) Les modifications de la substance blanche ne se limitent pas à une démyélinisation de type SEP, mais incluent également une vacuolisation et une pâleur myélinique diffuse. La dysfonction mitochondriale, la réponse auto-immune et le mimétisme moléculaire sont des mécanismes présumés de la démyélinisation inflammatoire. 1)
Les séquelles neurologiques vont de troubles légers à une évolution similaire à la SEP récurrente-rémittente.
7. Recherches récentes et perspectives futures (rapports en phase de recherche)
Une thérapie génique (lénadogène nolparvovec ; rAAV2/2-ND4) a été développée pour les patients LHON porteurs de la mutation MT-ND4. Elle introduit la protéine ND4 de type sauvage dans les mitochondries des cellules ganglionnaires rétiniennes par expression génique allotopique. 2)
L’étude REVERSE (2019) a rapporté une amélioration moyenne de la BCVA de -0,308 LogMAR dans les yeux injectés par voie intravitréenne et de -0,259 LogMAR dans les yeux non traités. 2) Ces résultats suggèrent un transfert immunologique vers l’œil controlatéral. Une analyse poolée (essais RESCUE, REVERSE, RESTORE, REFLECT) a montré une amélioration d’environ 21,5 lettres ETDRS par rapport au groupe témoin. L’administration bilatérale était plus efficace que l’unilatérale (environ 12 vs 8 lettres ETDRS), et une méta-analyse a montré que le rAAV2/2-ND4 était plus efficace que l’idébénone, les deux étant plus efficaces que l’évolution naturelle. 2)
Actuellement non approuvé par l’EMA et la FDA, son efficacité dans la maladie de Harding n’a pas été vérifiée. Un effet sur la composante LHON est attendu, mais l’impact sur la pathologie de la SEP n’est pas clair. 2)
Traitement par cellules souches mésenchymateuses (CSM)
Une technique consistant à administrer des CSM dérivées d’iPSC par voie intravitréenne pour transférer directement des mitochondries aux CGR est à l’étude. Les nanotubes de tunneling (TNT) dépendants de l’actine F médient le transfert. Une prévention de la diminution de la densité des CGR a été rapportée chez des souris Ndufs4 KO, mais aucun essai clinique n’a encore été mené. 3)
Une amélioration potentielle des potentiels évoqués visuels (PEV) dans la SEP a été suggérée, et son application chez les patients atteints de la maladie de Harding, de mauvais pronostic visuel, est à l’étude.
Le développement de biomarqueurs spécifiques à la maladie de Harding et des études prospectives sont nécessaires. Le traitement de cette maladie, qui associe les pathologies du LHON et de la SEP, doit être personnalisé, et une collaboration pluridisciplinaire est essentielle.
QLa thérapie génique peut-elle être utilisée pour la maladie de Harding ?
A
rAAV2/2-ND4 est une thérapie génique pour les patients LHON avec mutation MT-ND4, et son efficacité sur la composante LHON est attendue. 2)Cependant, son efficacité pour la maladie de Harding n’a pas été vérifiée et elle n’est actuellement pas approuvée par l’EMA ni la FDA. Son impact sur la pathologie de la SEP n’est pas clair, et une évaluation prudente est nécessaire pour son application à cette maladie.
Alorainy J, Alorfi Y, Karanjia R, Badeeb N. A Comprehensive Review of Leber Hereditary Optic Neuropathy and Its Association with Multiple Sclerosis-Like Phenotypes Known as Harding’s Disease. Eye and brain. 2024;16:17-24. doi:10.2147/EB.S470184. PMID:39100385; PMCID:PMC11296356.
Wong DCS, Makam R, Yu-Wai-Man P. Advanced therapies for inherited optic neuropathies. Eye (London, England). 2026;40(2):177-184. doi:10.1038/s41433-025-04109-1. PMID:41318849; PMCID:PMC12830909.
Hu JL, Hsu CC, Hsiao YJ, et al. Leber’s hereditary optic neuropathy: Update on the novel genes and therapeutic options. J Chin Med Assoc. 2024;87:12-16.
Sanders FWB, Votruba M. Outcomes of idebenone therapy for Leber hereditary optic neuropathy in a cohort of patients from Wales. Eye (Lond). 2025 Sep 17;39(16):2952-2957. doi:10.1038/s41433-025-03993-x. PMID:40962867; PMCID:PMC12583466.
Copiez le texte de l'article et collez-le dans l'assistant IA de votre choix.
Article copié dans le presse-papiers
Ouvrez un assistant IA ci-dessous et collez le texte copié dans la conversation.