Harding disease is a condition in which demyelinating neurological symptoms similar to multiple sclerosis (MS) coexist with Leber hereditary optic neuropathy (LHON) due to mitochondrial DNA (mtDNA) mutations. It was named after Harding et al., who reported in 1992 eight women with bilateral optic neuropathy and a family history of LHON, six of whom had neurological symptoms consistent with MS. 1)
A total of 88 cases have been reported in the literature. 1) Of these, 70.4% (62 cases) were women, with a male-to-female ratio of 2.38:1. 1) This contrasts with LHON, which predominantly affects men (93.1% male), suggesting that the female predominance in MS may play a role. The mean age of onset is 30.5 years. 1)
The prevalence of LHON is estimated at approximately 1 in 50,000 in Japan (with a total of about 4,000–5,000 patients nationwide), 1 in 31,000 in the UK, and 1 in 68,000 in Australia. 1) In 2015, LHON was designated as an intractable disease in Japan, and diagnostic criteria were established. The number of new cases is about 117 per year, with 47% occurring before the age of 30.
QHow rare is Harding disease?
A
Only 88 cases have been reported in the literature, making it an extremely rare disease. 1) Of these, 70.4% are women, and the mean age of onset is 30.5 years. Because it requires the coexistence of both LHON and MS, it is much rarer than either condition alone.
Visual loss starting with blurred vision: Begins with painless blurred vision, gradually worsening to severe visual loss and central scotoma.
Bilateral involvement: Usually affects both eyes. The interval until the second eye is affected is longer than in LHON, averaging 1.66 years. 1)
Multiple episodes of visual impairment: Unlike LHON, which typically has only two episodes, Harding disease causes multiple episodes of visual loss. 1)
No eye pain: Unlike optic neuritis in MS, it is not accompanied by eye pain. 1) This is an important point for differential diagnosis.
Extraocular symptoms: May be accompanied by postural tremor, peripheral neuropathy, movement disorders, cardiac arrhythmia, muscle weakness, and muscle disease.
Clinical Findings (Findings Confirmed by Physician Examination)
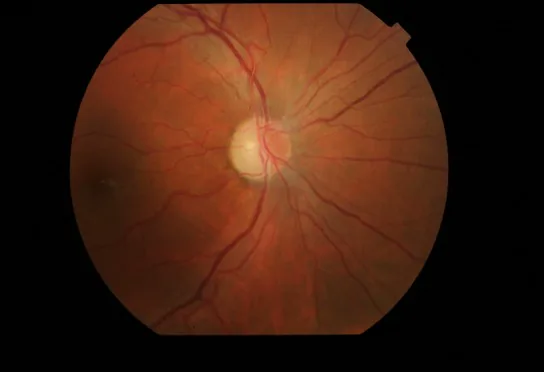
Optic disc redness and hyperemia: Accompanied by peripapillary RNFL edema.
Retinal capillary dilation: Capillary dilation (telangiectasia) and increased vascular tortuosity are observed.
No leakage on fluorescein angiography: No leakage of fluorescent dye from the hyperemic optic disc. This is an important distinguishing feature from optic neuritis.
OCT findings: Optic disc swelling is reportedly present before the onset of symptoms.
Chronic phase
Optic disc atrophy: Disc atrophy remains after inflammation subsides.
RNFL thinning: OCT shows thinning of the inner retinal layers, mainly the papillomacular bundle.
Decreased best-corrected visual acuity: Often remains around 0.01. Light perception is preserved.
Preservation of the pupillary light reflex: Compared to other optic nerve diseases, the pupillary light reflex is preserved or only mildly impaired.
Visual field: Presents with a dense central scotoma or centrocecal scotoma.
QHow does visual loss in Harding disease differ from that in LHON or optic neuritis in MS?
A
Differences from LHON include multiple episodes of visual loss and a longer interval before the second eye is affected (mean 1.66 years). 1) Differences from optic neuritis in MS include absence of eye pain and no leakage of dye from the optic disc on fluorescein angiography. These features provide important clues for differentiating Harding disease.
Harding disease is caused by pathogenic mutations in mitochondrial DNA (mtDNA). The three major mutations all encode complex I subunits, inducing apoptosis of retinal ganglion cells (RGCs) through impaired ATP synthesis and increased reactive oxygen species (ROS). 3)
The distribution of mutations in 88 cases of Harding disease is shown below.
Mutation
Gene
Proportion in Harding disease
m.11778G>A
MT-ND4
69.3% (61/88 cases)
m.14484T>C
MT-ND6
12.5%
m.3460G>A
MT-ND1
10.2%
Citation: Alorainy J et al. 20241)
The MT-ND4 mutation accounts for 90% of LHON cases in Asia and 70% in Europe.3) These three gene mutations also account for 95% of cases in Japan.
mtDNA is passed from mother to child (maternal inheritance). It is not passed on to the offspring of male patients.
Pathophysiological Hypothesis of Coexisting LHON and MS
The following three hypotheses have been proposed for the mechanism of coexistence of LHON and MS.1)
MS modification of LHON hypothesis: mtDNA mutations modify the phenotype of MS, causing painless, more severe, and irreversible atypical optic neuritis.
LHON manifestation due to MS predisposition: Genetic and environmental factors predisposing to MS in women promote the onset of LHON in asymptomatic mtDNA mutation carriers.
Incidental inflammation induction theory: In patients with LHON mtDNA mutations, an incidental inflammatory reaction is induced in the anterior visual pathway.
Smoking: May be involved as a risk factor for LHON onset.
Heavy alcohol consumption: May similarly be involved in the risk of onset.
Antituberculosis drugs (e.g., ethambutol): Clinical involvement has been noted.
QWhy is Harding disease more common in women?
A
LHON is male-predominant (93.1% male), but 70.4% of patients with Harding disease are female. This is related to the fact that MS is more common in women. It has been hypothesized that genetic and environmental factors predisposing to MS trigger LHON in asymptomatic mtDNA mutation carriers. 1) Estrogen signaling may also be involved in the pathogenesis of LHON. 2)
Harding disease is defined as “a patient who meets the MS diagnostic criteria and has a major LHON mutation” (Pfeffer et al.).
In Japan, the diagnostic criteria for LHON established in 2015 are applied.
Definite case: Meets the main signs (acute to subacute, bilateral, painless vision loss and central scotoma + at least one acute ophthalmoscopic abnormality).
Confirmed case: Main signs + mitochondrial gene missense mutation + no MRI retrobulbar optic nerve abnormality.
Suspected case: Clear maternal inheritance but no detectable genetic mutation.
Genetic testing: In Japan, testing for three mutations (m.3460, m.11778, m.14484) can be outsourced. If negative, referral to a core facility is required.
Exome sequencing: If mtDNA mutations are not identified, nuclear gene mutations such as DNAJC30 are detected.
MRI features: Findings that distinguish Harding disease from MS include decreased T2 lesion intensity and indistinct borders, absence of high signal on T1 lesions, and periventricular high signal areas (different from the typical Dawson’s fingers pattern). 1) 73% and 90% of LHON-MS and MS meet the McDonald criteria for spatial dissemination. 1)
Critical Flicker Frequency (CFF) / Pupillary Light Reflex Test: Maintained or only mildly decreased.
Screening considerations: In a screening of 1,666 MS patients, only 5 were positive for LHON mutations; routine screening is not recommended. Case-by-case decision is recommended, limited to patients with severe or bilateral visual field defects or maternal family history. 1)
No established treatment exists. The current approach is described below.
Idebenone (unapproved): A coenzyme Q10 derivative. It assists electron transport and may maintain or improve visual function. Clinical trials have been conducted in Japan, and some cases have reported improvement in visual function, but it is not approved domestically. Patients may personally import and take it orally.
Supplements: Coenzyme Q10, vitamin B complex, and vitamin C are used at the discretion of individual institutions.
Smoking cessation guidance: Since smoking is associated with the risk of developing LHON, patients are advised to quit smoking.
Genetic counseling: Recommended early for women who may transmit mtDNA mutations to their offspring. Provide information that male patients do not pass the mutation to their children, that spontaneous recovery is possible, and that the condition is recognized as an intractable disease.
Low vision care: Provide appropriate care and lifestyle guidance for patients with residual visual impairment.
While standard treatment has not been established, the following treatments have been attempted.
Intravenous methylprednisolone: Administration of 1 g/day for 3 days has been reported to result in mild to moderate visual improvement in some patients. 1)
Mitoxantrone: Intravenous administration of approximately 19.2 mg/month has been reported to improve visual acuity and neurological symptoms, but its use is limited due to severe side effects. 1)
Plasma exchange/cyclophosphamide: Some reports indicate subjective improvement in light perception and contrast sensitivity, but results are inconsistent. 1)
Immunomodulatory drugs: They stabilize clinical and MRI findings but cannot prevent the progression of visual impairment. Inflammatory rebound after discontinuation of natalizumab has been reported. 1)
QWhat treatments are used for Harding disease in Japan?
A
There is no established treatment, and symptomatic therapy is the mainstay. Some patients personally import idebenone, which is not approved domestically, for oral use, but evidence of its efficacy in Harding disease is lacking. 4)Currently, supplements such as CoQ10 and vitamin B complex, smoking cessation guidance, low vision care, and genetic counseling are provided.
The basic mechanism of LHON originates from impairment of complex I (mitochondrial respiratory chain) subunits due to mtDNA mutations. Impaired electron transport reduces ATP synthesis and simultaneously accumulates reactive oxygen species (ROS). This induces apoptosis of RGCs, leading to optic atrophy. 3)
There are differences in visual recovery rates depending on the mutation. The visual recovery rate for the MT-ND4 mutation is 4–25%, which is lower than that for MT-ND1 and MT-ND6 mutations. 3) In Japan, the most common mt11778 (MT-ND4) mutation has a visual improvement rate of only a few percent. The mt14484 (MT-ND6) mutation has the highest improvement rate.
The higher penetrance of LHON in males suggests the involvement of an X-linked nuclear gene. 3)
PRICKLE3 (X chromosome Xp11.23): Regulates the function of ATP synthase (complex V). 3)
YARS2 mutation: Impairs the function of electron transport chain complexes I, III, and IV. 3)
In the histopathology of LHON-MS, T cells and activated macrophages/microglia mediate tissue damage. The presence of inflammatory cells within LHON lesions is unusual and suggests an early immunological mechanism. 1) White matter changes involve not only MS-like demyelination but also vacuolation and diffuse myelin pallor. Mitochondrial dysfunction, autoimmune response, and molecular mimicry are hypothesized mechanisms of inflammatory demyelination. 1)
Neurological sequelae range from mild impairment to a course resembling relapsing-remitting MS.
7. Latest Research and Future Perspectives (Reports under Investigation)
Gene therapy (lenadogene nolparvovec; rAAV2/2-ND4) has been developed for LHON patients with MT-ND4 mutations. It introduces wild-type ND4 protein into the mitochondria of RGCs through allotopic gene expression. 2)
In the REVERSE study (2019), mean BCVA improvement of -0.308 LogMAR was reported in the injected eye and -0.259 LogMAR in the untreated eye. 2) This result suggested immunological transfer to the contralateral eye. Pooled analysis (RESCUE, REVERSE, RESTORE, REFLECT trials) showed approximately 21.5 ETDRS letter improvement compared to the control group. Bilateral administration was more effective than unilateral (approximately 12 vs 8 ETDRS letters), and meta-analysis showed rAAV2/2-ND4 was more effective than idebenone, both being more effective than natural history. 2)
Currently not approved by EMA or FDA, and efficacy for Harding disease has not been verified. While effects on the LHON component are expected, the impact on MS pathology is unclear. 2)
A method of intravitreal administration of iPSC-derived MSCs to directly transfer mitochondria to RGCs is being studied. F-actin-dependent tunneling nanotubes (TNTs) mediate the transfer. Prevention of RGC density reduction has been reported in Ndufs4 KO mice, but clinical trials have not yet been conducted. 3)
It has been suggested that 4-aminopyridine may improve visual evoked potentials (VEP) in MS, and its application to Harding disease patients with poor visual prognosis is being considered.
Development of biomarkers specific to Harding disease and prospective studies are needed. Treatment for this disease, which has the dual pathology of LHON and MS, should be individualized, and multidisciplinary collaboration is essential.
QCan gene therapy be used for Harding disease?
A
rAAV2/2-ND4 is a gene therapy for LHON patients with MT-ND4 mutations and is expected to be effective for the LHON component. 2) However, its efficacy for Harding disease has not been verified, and it is currently not approved by the EMA or FDA. Its impact on MS pathology is also unclear, and careful consideration is needed for its application to this disease.
Alorainy J, Alorfi Y, Karanjia R, Badeeb N. A Comprehensive Review of Leber Hereditary Optic Neuropathy and Its Association with Multiple Sclerosis-Like Phenotypes Known as Harding’s Disease. Eye and brain. 2024;16:17-24. doi:10.2147/EB.S470184. PMID:39100385; PMCID:PMC11296356.
Wong DCS, Makam R, Yu-Wai-Man P. Advanced therapies for inherited optic neuropathies. Eye (London, England). 2026;40(2):177-184. doi:10.1038/s41433-025-04109-1. PMID:41318849; PMCID:PMC12830909.
Hu JL, Hsu CC, Hsiao YJ, et al. Leber’s hereditary optic neuropathy: Update on the novel genes and therapeutic options. J Chin Med Assoc. 2024;87:12-16.
Sanders FWB, Votruba M. Outcomes of idebenone therapy for Leber hereditary optic neuropathy in a cohort of patients from Wales. Eye (Lond). 2025 Sep 17;39(16):2952-2957. doi:10.1038/s41433-025-03993-x. PMID:40962867; PMCID:PMC12583466.
Copy the article text and paste it into your preferred AI assistant.
Article copied to clipboard
Open an AI assistant below and paste the copied text into the chat box.