La enfermedad de Harding es una afección en la que los síntomas neurológicos desmielinizantes similares a la esclerosis múltiple (EM) coexisten con la neuropatía óptica hereditaria de Leber (NOHL) debida a mutaciones en el ADN mitocondrial (ADNmt). Recibe su nombre de Harding et al., quienes en 1992 reportaron ocho mujeres con neuropatía óptica bilateral y antecedentes familiares de NOHL, seis de las cuales presentaban síntomas neurológicos compatibles con EM. 1)
Se han reportado un total de 88 casos en la literatura. 1) El 70.4% (62 casos) eran mujeres, con una proporción hombre:mujer de 2.38:1. 1) Esto contrasta con la NOHL, que afecta predominantemente a hombres (93.1% hombres), lo que sugiere que la predisposición femenina en la EM puede desempeñar un papel. La edad media de inicio es de 30.5 años. 1)
La prevalencia de la NOHL se estima en aproximadamente 1/50,000 en Japón (con un total de unos 4,000–5,000 pacientes en todo el país), 1/31,000 en el Reino Unido y 1/68,000 en Australia. 1) En 2015, la NOHL fue designada como enfermedad intratable en Japón y se establecieron criterios diagnósticos. El número de nuevos casos es de aproximadamente 117 por año, con un 47% que ocurren antes de los 30 años.
Q¿Qué tan rara es la enfermedad de Harding?
A
Solo se han reportado 88 casos en la literatura, por lo que es una enfermedad extremadamente rara. 1) El 70.4% son mujeres y la edad media de inicio es de 30.5 años. Debido a que requiere la coexistencia de NOHL y EM, es mucho más rara que cualquiera de las dos condiciones por separado.
Pérdida visual que comienza con visión borrosa: Comienza con visión borrosa indolora, empeorando gradualmente hasta una pérdida visual severa y escotoma central.
Afectación bilateral: Generalmente afecta ambos ojos. El intervalo hasta que el segundo ojo se ve afectado es más largo que en LHON, con un promedio de 1.66 años. 1)
Múltiples episodios de deterioro visual: A diferencia de LHON, que típicamente tiene solo dos episodios, la enfermedad de Harding causa múltiples episodios de pérdida visual. 1)
Sin dolor ocular: A diferencia de la neuritis óptica en la EM, no se acompaña de dolor ocular. 1) Este es un punto importante para el diagnóstico diferencial.
Síntomas extraoculares: Puede acompañarse de temblor postural, neuropatía periférica, trastornos del movimiento, arritmia cardíaca, debilidad muscular y enfermedad muscular.
Hallazgos clínicos (hallazgos confirmados por el médico en el examen)
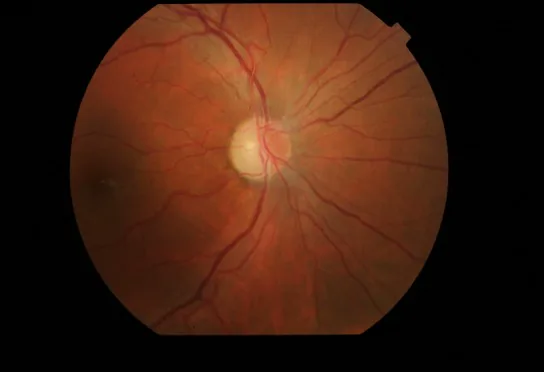
Enrojecimiento e hiperemia del disco óptico: Acompañado de edema de la capa de fibras nerviosas peripapilar.
Dilatación capilar retiniana: Se observa dilatación capilar (telangiectasia) y aumento de la tortuosidad vascular.
Ausencia de fuga en la angiografía fluoresceínica: No se observa fuga de colorante fluorescente desde el disco óptico hiperémico. Este es un punto importante de diferenciación con la neuritis óptica.
Hallazgos en OCT: Se reporta que la hinchazón del disco óptico está presente antes del inicio de los síntomas.
Fase crónica
Atrofia del disco óptico: La atrofia del disco persiste después de que la inflamación se resuelve.
Adelgazamiento de la RNFL: La OCT muestra adelgazamiento de las capas internas de la retina, principalmente del haz papilomacular.
Disminución de la mejor agudeza visual corregida: A menudo se mantiene alrededor de 0.01. La percepción de la luz se conserva.
Preservación del reflejo pupilar a la luz: En comparación con otras enfermedades del nervio óptico, el reflejo pupilar a la luz se conserva o solo se altera levemente.
Campo visual: Presenta un escotoma central denso o un escotoma centrocecal.
Q¿En qué se diferencia la pérdida de visión en la enfermedad de Harding de la de la LHON o la neuritis óptica en la EM?
A
Las diferencias con la LHON incluyen múltiples episodios de pérdida de visión y un intervalo más largo antes de que el segundo ojo se vea afectado (media de 1.66 años). 1) Las diferencias con la neuritis óptica en la EM incluyen la ausencia de dolor ocular y la ausencia de fuga de colorante del disco óptico en la angiografía con fluoresceína. Estas características proporcionan pistas importantes para diferenciar la enfermedad de Harding.
La enfermedad de Harding es causada por mutaciones patogénicas en el ADN mitocondrial (ADNmt). Las tres mutaciones principales codifican subunidades del complejo I, induciendo apoptosis de las células ganglionares de la retina (CGR) a través de la alteración de la síntesis de ATP y el aumento de especies reactivas de oxígeno (ERO). 3)
La distribución de mutaciones en 88 casos de enfermedad de Harding se muestra a continuación.
Mutación
Gen
Proporción en enfermedad de Harding
m.11778G>A
MT-ND4
69.3% (61/88 casos)
m.14484T>C
MT-ND6
12.5%
m.3460G>A
MT-ND1
10.2%
Citación: Alorainy J et al. 20241)
La mutación MT-ND4 representa el 90% de los casos de LHON en Asia y el 70% en Europa.3) Estas tres mutaciones genéticas también representan el 95% de los casos en Japón.
El mtDNA se transmite de madre a hijo (herencia materna). No se transmite a la descendencia de pacientes varones.
Hipótesis fisiopatológica de la coexistencia de LHON y EM
Se han propuesto las siguientes tres hipótesis para el mecanismo de coexistencia de LHON y EM.1)
Hipótesis de modificación de LHON por EM: las mutaciones del mtDNA modifican el fenotipo de la EM, causando neuritis óptica atípica indolora, más grave e irreversible.
Teoría de manifestación de LHON por predisposición a EM: Los factores genéticos y ambientales que predisponen a EM en mujeres promueven la aparición de LHON en portadores asintomáticos de mutaciones del mtDNA.
Teoría de inducción inflamatoria incidental: En pacientes con mutaciones del mtDNA de LHON, se induce una reacción inflamatoria incidental en la vía visual anterior.
Tabaquismo: Puede estar implicado como factor de riesgo para el desarrollo de LHON.
Consumo excesivo de alcohol: Puede estar implicado de manera similar en el riesgo de aparición.
Fármacos antituberculosos (como etambutol): Se ha señalado su implicación clínica.
Q¿Por qué la enfermedad de Harding es más común en mujeres?
A
La LHON es predominante en hombres (93.1% hombres), pero el 70.4% de los pacientes con enfermedad de Harding son mujeres. Esto se relaciona con el hecho de que la EM es más común en mujeres. Se ha planteado la hipótesis de que factores genéticos y ambientales que predisponen a la EM desencadenan la LHON en portadores asintomáticos de mutaciones en el ADNmt. 1) También se ha sugerido que la señalización de estrógenos podría estar involucrada en la patogenia de la LHON. 2)
La enfermedad de Harding se define como “un paciente que cumple los criterios diagnósticos de EM y tiene una mutación importante de LHON” (Pfeffer et al.).
En Japón se aplican los criterios diagnósticos de LHON establecidos en 2015.
Caso definitivo: Cumple los signos principales (pérdida visual aguda a subaguda, bilateral, indolora y escotoma central + al menos una anomalía oftalmoscópica en la fase aguda).
Caso confirmado: Signos principales + mutación de sentido erróneo en el gen mitocondrial + sin anomalía del nervio óptico retrobulbar en la RM.
Caso sospechoso: Herencia materna clara pero sin mutación genética detectable.
Pruebas genéticas: En Japón, las pruebas para tres mutaciones (m.3460, m.11778, m.14484) se pueden subcontratar. Si son negativas, se requiere derivación a un centro de referencia.
Secuenciación del exoma: si no se identifican mutaciones en el ADNmt, se detectan mutaciones en genes nucleares como DNAJC30.
Características de la RM: Los hallazgos que distinguen la enfermedad de Harding de la EM incluyen disminución de la intensidad de la lesión en T2 y bordes indistintos, ausencia de señal alta en lesiones T1 y áreas de señal alta periventriculares (diferentes del patrón típico de dedos de Dawson). 1) El 73% y el 90% de LHON-EM y EM cumplen con los criterios de McDonald para diseminación espacial. 1)
Frecuencia crítica de fusión (CFF) / Prueba de reflejo pupilar a la luz: Se mantiene o solo disminuye levemente.
Consideraciones de cribado: En un cribado de 1.666 pacientes con EM, solo 5 fueron positivos para mutaciones LHON; no se recomienda el cribado rutinario. Se recomienda una decisión caso por caso, limitada a pacientes con defectos del campo visual graves o bilaterales o antecedentes familiares maternos. 1)
No existe un tratamiento establecido. A continuación se describe el enfoque actual.
Idebenona (no aprobado): Derivado de la coenzima Q10. Asiste en el transporte de electrones y puede mantener o mejorar la función visual. Se han realizado ensayos clínicos en Japón y algunos casos han reportado mejoría, pero no está aprobado en el país. Los pacientes pueden importarlo personalmente y tomarlo por vía oral.
Suplementos: Coenzima Q10, complejo vitamínico B y vitamina C se utilizan según el criterio de cada institución.
Consejo para dejar de fumar: Dado que fumar está relacionado con el riesgo de desarrollar LHON, se recomienda a los pacientes que dejen de fumar.
Asesoramiento genético: Se recomienda tempranamente para mujeres que pueden transmitir la mutación del mtDNA a su descendencia. Proporcionar información de que los pacientes varones no transmiten la mutación a sus hijos, que existe la posibilidad de recuperación espontánea y que la enfermedad está reconocida como enfermedad rara.
Cuidados de baja visión: Proporcionar cuidados adecuados y orientación sobre la vida diaria a pacientes con discapacidad visual residual.
Informes de tratamiento en el extranjero (complemento)
En ausencia de un tratamiento estándar establecido, se han intentado los siguientes tratamientos.
Metilprednisolona intravenosa: Se ha informado que la administración de 1 g/día durante 3 días produce una mejoría visual leve a moderada en algunos pacientes. 1)
Mitoxantrona: La administración intravenosa de aproximadamente 19.2 mg/mes ha mostrado mejoría en la agudeza visual y síntomas neurológicos, pero su uso es limitado debido a efectos secundarios graves. 1)
Plasmaféresis/ciclofosfamida: Algunos informes indican mejoría subjetiva en la percepción de la luz y la sensibilidad al contraste, pero los resultados no son consistentes. 1)
Fármacos inmunomoduladores: Proporcionan estabilidad clínica y en los hallazgos de RM, pero no pueden prevenir la progresión del deterioro visual. Se ha reportado un rebote inflamatorio tras la suspensión de natalizumab. 1)
Q¿Qué tratamientos se utilizan para la enfermedad de Harding en Japón?
A
No existe un tratamiento establecido, y la terapia sintomática es el pilar. Algunos pacientes importan personalmente idebenona, no aprobada a nivel nacional, para uso oral, pero la evidencia de su eficacia en la enfermedad de Harding es escasa. 4)Actualmente se proporcionan suplementos como CoQ10 y complejo vitamínico B, consejos para dejar de fumar, cuidado de baja visión y asesoramiento genético.
6. Fisiopatología y mecanismo detallado de la enfermedad
El mecanismo básico de LHON se origina por la alteración de las subunidades del complejo I (cadena respiratoria mitocondrial) debido a mutaciones en el ADNmt. El deterioro del transporte de electrones reduce la síntesis de ATP y simultáneamente acumula especies reactivas de oxígeno (ROS). Esto induce la apoptosis de las CGR, lo que lleva a la atrofia óptica. 3)
Existen diferencias en las tasas de recuperación visual según la mutación. La tasa de recuperación visual para la mutación MT-ND4 es del 4 al 25%, inferior a la de las mutaciones MT-ND1 y MT-ND6. 3) En Japón, la mutación más común, mt11778 (MT-ND4), tiene una tasa de mejora visual de solo unos pocos por ciento. La mutación mt14484 (MT-ND6) tiene la tasa de mejora más alta.
La mayor penetrancia de LHON en varones sugiere la participación de un gen nuclear ligado al cromosoma X. 3)
PRICKLE3 (cromosoma X Xp11.23): Regula la función de la ATP sintasa (complejo V). 3)
Mutación YARS2: Altera la función de los complejos I, III y IV de la cadena de transporte de electrones. 3)
Mutación DNAJC30 (c.152A>G): Altera los mecanismos de reparación del complejo I, causando LHON autosómico recesivo. 3)
En la histopatología de LHON-MS, los linfocitos T y los macrófagos/microglía activados median el daño tisular. La presencia de células inflamatorias dentro de las lesiones de LHON es inusual y sugiere un mecanismo inmunológico temprano. 1) Los cambios en la sustancia blanca incluyen no solo desmielinización similar a la EM, sino también vacuolización y palidez difusa de la mielina. La disfunción mitocondrial, la respuesta autoinmune y el mimetismo molecular son mecanismos hipotéticos de la desmielinización inflamatoria. 1)
Las secuelas neurológicas varían desde discapacidad leve hasta un curso similar a la EM remitente-recurrente.
7. Investigación más reciente y perspectivas futuras (Informes en fase de investigación)
Se ha desarrollado una terapia génica (lenadogeno nolparvovec; rAAV2/2-ND4) para pacientes con LHON con mutación MT-ND4. Introduce la proteína ND4 de tipo salvaje en las mitocondrias de las CGR mediante expresión génica alotópica. 2)
En el estudio REVERSE (2019), se informó una mejora media de la AVCC de -0,308 LogMAR en el ojo inyectado intravítreo y de -0,259 LogMAR en el ojo no tratado. 2) Este resultado sugirió una transferencia inmunológica al ojo contralateral. El análisis combinado (ensayos RESCUE, REVERSE, RESTORE, REFLECT) mostró una mejora de aproximadamente 21,5 letras ETDRS en comparación con el grupo de control. La administración bilateral fue más efectiva que la unilateral (aproximadamente 12 vs 8 letras ETDRS), y el metanálisis mostró que rAAV2/2-ND4 fue más efectivo que idebenona, siendo ambos más efectivos que la historia natural. 2)
Actualmente no aprobado por la EMA ni la FDA, y no se ha verificado la eficacia para la enfermedad de Harding. Si bien se esperan efectos sobre el componente LHON, el impacto sobre la patología de la EM no está claro. 2)
Se está investigando un método de administración intravítrea de MSC derivadas de iPSC para transferir directamente mitocondrias a las CGR. Los nanotubos de túnel (TNT) dependientes de F-actina median la transferencia. Se ha informado la prevención de la disminución de la densidad de CGR en ratones Ndufs4 KO, pero aún no se han realizado ensayos clínicos. 3)
Se ha sugerido que la 4-aminopiridina puede mejorar los potenciales evocados visuales (PEV) en la EM, y se está considerando su aplicación en pacientes con enfermedad de Harding con mal pronóstico visual.
Se necesita el desarrollo de biomarcadores específicos para la enfermedad de Harding y estudios prospectivos. El tratamiento para esta enfermedad, que presenta la patología dual de LHON y EM, debe individualizarse, y la colaboración multidisciplinaria es esencial.
Q¿Se puede usar la terapia génica para la enfermedad de Harding?
A
rAAV2/2-ND4 es una terapia génica para pacientes con LHON con mutaciones en MT-ND4 y se espera que sea efectiva para el componente de LHON. 2) Sin embargo, su eficacia para la enfermedad de Harding no ha sido verificada y actualmente no está aprobada por la EMA ni la FDA. Su impacto en la patología de la EM tampoco está claro, y se necesita una consideración cuidadosa para su aplicación en esta enfermedad.
Alorainy J, Alorfi Y, Karanjia R, Badeeb N. A Comprehensive Review of Leber Hereditary Optic Neuropathy and Its Association with Multiple Sclerosis-Like Phenotypes Known as Harding’s Disease. Eye and brain. 2024;16:17-24. doi:10.2147/EB.S470184. PMID:39100385; PMCID:PMC11296356.
Wong DCS, Makam R, Yu-Wai-Man P. Advanced therapies for inherited optic neuropathies. Eye (London, England). 2026;40(2):177-184. doi:10.1038/s41433-025-04109-1. PMID:41318849; PMCID:PMC12830909.
Hu JL, Hsu CC, Hsiao YJ, et al. Leber’s hereditary optic neuropathy: Update on the novel genes and therapeutic options. J Chin Med Assoc. 2024;87:12-16.
Sanders FWB, Votruba M. Outcomes of idebenone therapy for Leber hereditary optic neuropathy in a cohort of patients from Wales. Eye (Lond). 2025 Sep 17;39(16):2952-2957. doi:10.1038/s41433-025-03993-x. PMID:40962867; PMCID:PMC12583466.
Copia el texto del artículo y pégalo en el asistente de IA que prefieras.
Artículo copiado al portapapeles
Abre un asistente de IA abajo y pega el texto copiado en el chat.