La malattia di Harding è una condizione in cui sintomi neurologici demielinizzanti simili alla sclerosi multipla (SM) coesistono con una mutazione del DNA mitocondriale (mtDNA) della neuropatia ottica ereditaria di Leber (LHON). Prende il nome dal rapporto del 1992 di Harding et al., che descrisse otto donne con neuropatia ottica bilaterale e storia familiare di LHON (di cui sei con sintomi neurologici compatibili con SM). 1)
In letteratura sono stati riportati finora un totale di 88 casi. 1) Il 70,4% (62 casi) dei pazienti sono donne, con un rapporto maschi:femmine di 2,38:1. 1) Ciò contrasta con la LHON, che è più comune negli uomini (93,1%), e suggerisce il coinvolgimento di una predisposizione femminile alla SM. L’età media di insorgenza è di 30,5 anni. 1)
La prevalenza della LHON è stimata in circa 1/50.000 in Giappone (numero totale di pazienti nel paese stimato circa 4.000-5.000), 1/31.000 nel Regno Unito e 1/68.000 in Australia. 1) Nel 2015, la LHON è stata designata come malattia rara in Giappone e sono stati stabiliti criteri diagnostici. Il numero di nuovi casi è di circa 117 all’anno, con il 47% che si verifica entro i 30 anni.
QQuanto è rara la malattia di Harding?
A
In letteratura sono stati riportati solo 88 casi, rendendola una malattia estremamente rara. 1) Il 70,4% dei pazienti sono donne e l’età media è di 30,5 anni. Poiché richiede la coesistenza di LHON e SM, è molto meno comune della sola LHON o SM.
Riduzione dell’acuità visiva che inizia con offuscamento : inizia con un offuscamento indolore, peggiora gradualmente fino a una grave riduzione dell’acuità visiva e scotoma centrale.
Coinvolgimento bilaterale : di solito colpisce entrambi gli occhi. L’intervallo prima del coinvolgimento del secondo occhio è più lungo rispetto alla LHON, con una media di 1,66 anni. 1)
Episodi multipli di disturbi visivi : mentre la LHON di solito ne causa solo due, la malattia di Harding provoca episodi multipli di riduzione dell’acuità visiva. 1)
Senza dolore oculare : A differenza della neurite ottica nella SM, non è accompagnata da dolore oculare. 1) Questo è un punto chiave per la diagnosi differenziale.
Sintomi extraoculari : Può essere accompagnato da tremore posturale, neuropatia periferica, disturbi motori, aritmia cardiaca, debolezza muscolare, malattie muscolari.
Reperti clinici (reperti osservati dal medico durante l’esame)
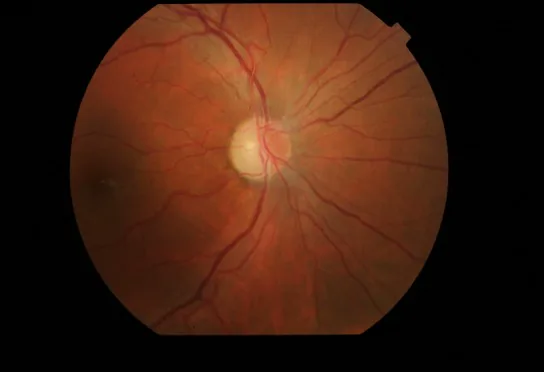
Arrossamento e iperemia della papilla ottica : Con edema peripapillare dello strato di fibre nervose retiniche (RNFL).
Teleangectasie retiniche : Dilatazione dei capillari (teleangectasia) e aumento della tortuosità vascolare.
Assenza di perdita all’angiografia con fluoresceina : Non si osserva perdita di colorante fluorescente dalla papilla ottica arrossata. Questo è un importante punto di differenziazione dalla neurite ottica.
Risultati OCT : Si ritiene che il gonfiore della papilla ottica sia osservabile prima dell’insorgenza dei sintomi.
Fase cronica
Atrofia della papilla ottica : Dopo la risoluzione dell’infiammazione, persiste l’atrofia papillare.
Assottigliamento della RNFL : L’OCT mostra un assottigliamento degli strati interni della retina, principalmente a livello del fascio papillomaculare.
Riduzione della migliore acuità visiva corretta : Spesso rimane intorno a 0,01. La percezione luminosa è preservata.
Conservazione del riflesso pupillare alla luce: rispetto ad altre malattie del nervo ottico, il riflesso pupillare alla luce è conservato o solo lievemente compromesso.
Campo visivo: presenta uno scotoma centrale denso o uno scotoma centrocecale (centrocecal scotoma).
QIn che modo la riduzione della vista nella malattia di Harding differisce da quella nella LHON o nella neurite ottica da sclerosi multipla?
A
La differenza rispetto alla LHON è la presenza di multipli episodi di riduzione della vista e un intervallo più lungo prima del coinvolgimento del secondo occhio (media 1,66 anni). 1) La differenza rispetto alla neurite ottica da sclerosi multipla è l’assenza di dolore oculare e l’assenza di perdita di colorante dalla papilla ottica all’angiografia con fluoresceina. Queste caratteristiche sono indizi importanti per la diagnosi differenziale della malattia di Harding.
La malattia di Harding è causata da mutazioni patogene del DNA mitocondriale (mtDNA). Le tre mutazioni principali codificano tutte per subunità del complesso I, inducendo l’apoptosi delle cellule gangliari retiniche (RGC) attraverso un deficit di sintesi di ATP e un aumento delle specie reattive dell’ossigeno (ROS). 3)
La distribuzione delle mutazioni in 88 casi di malattia di Harding è mostrata di seguito.
Mutazione
Gene
Proporzione nella malattia di Harding
m.11778G>A
MT-ND4
69,3% (61/88 casi)
m.14484T>C
MT-ND6
12,5 %
m.3460G>A
MT-ND1
10,2 %
Citazione: Alorainy J et al. 20241)
La mutazione MT-ND4 rappresenta il 90% dei casi di LHON in Asia e il 70% in Europa.3) Anche nei pazienti giapponesi, queste tre mutazioni genetiche rappresentano il 95% dei casi.
Il mtDNA viene trasmesso dalla madre al figlio (eredità materna). Non viene trasmesso alla prole dei pazienti di sesso maschile.
Per il meccanismo di coesistenza di LHON e SM sono state proposte le seguenti tre ipotesi.1)
Ipotesi di modifica della LHON da parte della SM: la mutazione del mtDNA modifica il fenotipo della SM, causando una neurite ottica atipica, indolore, più grave e irreversibile.
Teoria della manifestazione della LHON dovuta a predisposizione alla SM: Nelle donne, fattori genetici e ambientali che predispongono alla SM favoriscono l’insorgenza della LHON in portatori asintomatici di mutazioni del mtDNA.
Teoria dell’induzione infiammatoria accidentale: Nei pazienti con mutazioni del mtDNA della LHON, viene indotta una reazione infiammatoria accidentale nelle vie visive anteriori.
Fumo: Può essere coinvolto come fattore di rischio per lo sviluppo della LHON.
Consumo eccessivo di alcol: Può essere ugualmente coinvolto nel rischio di sviluppo.
Farmaci antitubercolari (etambutolo, ecc.): È stato segnalato un coinvolgimento clinico.
QPerché la malattia di Harding è più comune nelle donne?
A
La LHON è predominante negli uomini (93,1%), mentre nella malattia di Harding il 70,4% dei pazienti sono donne. Ciò è correlato al fatto che la sclerosi multipla (SM) è più comune nelle donne. È stata ipotizzata una teoria secondo cui fattori genetici e ambientali predisponenti alla SM scatenano la LHON in portatori asintomatici di mutazioni del mtDNA. 1) È stato anche suggerito che la segnalazione degli estrogeni possa essere coinvolta nella patogenesi della LHON. 2)
La malattia di Harding è definita come «un paziente che soddisfa i criteri diagnostici per la SM e presenta una mutazione LHON maggiore» (Pfeffer et al.).
In Giappone vengono applicati i criteri diagnostici per la LHON stabiliti nel 2015.
Caso confermato : soddisfa i criteri principali (riduzione acuta o subacuta, bilaterale, indolore della vista con scotoma centrale + almeno un’anomalia oftalmoscopica in fase acuta).
Caso certo : segni principali + mutazione missenso del gene mitocondriale + RM senza anomalie del nervo ottico retrobulbare
Caso sospetto : eredità materna evidente ma mutazione genetica non rilevabile
Esclusione della sindrome da preeccitazione cardiaca
Test genetico: in Giappone è possibile eseguire esternamente i test per le tre mutazioni m.3460, m.11778 e m.14484. In caso di negatività, è necessario rivolgersi a un centro di riferimento.
Sequenziamento dell’esoma : se non viene identificata una mutazione del mtDNA, rilevare mutazioni di geni nucleari come DNAJC30.
Caratteristiche RM: I reperti che distinguono la malattia di Harding dalla SM includono una ridotta luminosità e margini sfumati delle lesioni T2, l’assenza di segnale elevato nelle lesioni T1 e un’area di segnale elevato intorno al corno anteriore (diversa dal tipico pattern delle dita di Dawson). 1) Il 73% e il 90% dei casi di LHON-SM e SM soddisfano i criteri di McDonald per la disseminazione spaziale. 1)
Frequenza critica di fusione (CFF) e test del riflesso pupillare alla luce : conservata o lievemente ridotta.
Considerazioni sullo screening : In uno screening su 1.666 pazienti con SM, solo 5 sono risultati positivi alla mutazione LHON, pertanto lo screening di routine non è raccomandato. Si raccomanda una decisione caso per caso, limitata a pazienti con grave o bilaterale difetto del campo visivo o anamnesi familiare materna. 1)
Non esiste un trattamento consolidato. Di seguito sono riportate le opzioni attuali.
Idebenone (non approvato) : Derivato del coenzima Q10. Aiuta il trasporto degli elettroni e può mantenere o migliorare la funzione visiva. In Giappone sono stati condotti studi clinici e in alcuni casi è stato riportato un miglioramento visivo, ma il farmaco non è approvato in Giappone. I pazienti possono importarlo per uso personale.
Integratori : Coenzima Q10, vitamine del gruppo B, vitamina C, ecc., sono utilizzati a discrezione di ogni struttura.
Consigli per smettere di fumare: Poiché il fumo è un fattore di rischio per lo sviluppo di LHON, si consiglia di smettere di fumare.
Consulenza genetica: raccomandata precocemente per le donne che potrebbero trasmettere una mutazione del mtDNA alla prole. Informare che i pazienti di sesso maschile non trasmettono la mutazione ai figli, che esistono casi di guarigione spontanea e che la malattia è riconosciuta come malattia rara.
Cura della bassa visione : fornire cure appropriate e consigli sulla vita ai pazienti con deficit visivo residuo.
In assenza di un trattamento standard consolidato, vengono tentati i seguenti trattamenti.
Metilprednisolone endovena: somministrazione di 1 g/die per 3 giorni, in alcuni pazienti è stato riportato un miglioramento visivo da lieve a moderato. 1)
Mitoxantrone : circa 19,2 mg/mese per via endovenosa ha mostrato miglioramento della vista e dei sintomi neurologici, ma l’uso è limitato a causa di gravi effetti collaterali. 1)
Plasmaferesi e ciclofosfamide : in alcuni casi è stato riportato un miglioramento soggettivo della percezione della luce e della sensibilità al contrasto, ma non coerente. 1)
Immunomodulatori: portano a una stabilizzazione dei reperti clinici e di risonanza magnetica, ma non possono arrestare la progressione del deficit visivo. Sono stati riportati casi di rebound infiammatorio dopo la sospensione di natalizumab. 1)
QQuale trattamento viene eseguito in Giappone per la malattia di Harding?
A
Non esiste un trattamento standardizzato; la terapia è principalmente sintomatica. Alcuni pazienti importano personalmente l’idebenone, non approvato in Giappone, per assumerlo per via orale, ma le prove di efficacia nella malattia di Harding sono scarse. 4)Attualmente vengono forniti integratori come CoQ10 e vitamine del gruppo B, consigli per smettere di fumare, cure per la bassa visione e consulenza genetica.
Il meccanismo di base della LHON origina da una mutazione del mtDNA che causa un danno alle subunità del complesso I (catena respiratoria mitocondriale). Il difetto nel trasporto degli elettroni riduce la sintesi di ATP e accumula contemporaneamente specie reattive dell’ossigeno (ROS). Ciò induce l’apoptosi delle cellule gangliari retiniche (RGC), portando all’atrofia del nervo ottico. 3)
I tassi di recupero visivo variano a seconda della mutazione: la mutazione MT-ND4 ha un tasso di recupero del 4-25%, inferiore a quello delle mutazioni MT-ND1 e MT-ND6. 3)In Giappone, la mutazione più comune mt11778 (MT-ND4) ha un tasso di miglioramento della funzione visiva di solo pochi punti percentuali. La mutazione mt14484 (MT-ND6) ha il tasso di recupero più alto.
La maggiore penetranza della LHON nei maschi suggerisce il coinvolgimento di geni nucleari legati al cromosoma X. 3)
PRICKLE3 (cromosoma X, Xp11.23): regola la funzione dell’ATP sintasi (complesso V). 3)
Mutazione YARS2 : danneggia la funzione dei complessi I, III e IV della catena di trasporto degli elettroni. 3)
Mutazione DNAJC30 (c.152A>G) : danneggia il meccanismo di riparazione del complesso I, causando LHON autosomica recessiva. 3)
L’istopatologia della LHON-MS mostra che linfociti T e macrofagi/microglia attivati mediano il danno tissutale. La presenza di cellule infiammatorie nelle lesioni LHON è insolita e suggerisce un meccanismo immunologico precoce. 1) Le alterazioni della sostanza bianca non sono solo demielinizzazione simile alla SM, ma contribuiscono anche vacuolizzazione e pallore mielinico diffuso. Disfunzione mitocondriale, risposta autoimmune e mimetismo molecolare sono ipotizzati come meccanismi della demielinizzazione infiammatoria. 1)
Le sequele neurologiche vanno da disturbi lievi a un decorso simile alla SM recidivante-remittente.
7. Ricerche recenti e prospettive future (rapporti in fase di ricerca)
È stata sviluppata una terapia genica (lenadogene nolparvovec; rAAV2/2-ND4) per i pazienti con LHON portatori della mutazione MT-ND4. Introduce la proteina ND4 wild-type nei mitocondri delle cellule gangliari retiniche mediante espressione genica allotopica. 2)
Lo studio REVERSE (2019) ha riportato un miglioramento medio della BCVA di -0,308 LogMAR negli occhi con iniezione intravitreale e di -0,259 LogMAR negli occhi non trattati. 2) Questi risultati suggeriscono un trasferimento immunologico all’occhio controlaterale. Un’analisi aggregata (studi RESCUE, REVERSE, RESTORE, REFLECT) ha mostrato un miglioramento di circa 21,5 lettere ETDRS rispetto al gruppo di controllo. La somministrazione bilaterale è stata più efficace di quella unilaterale (circa 12 vs 8 lettere ETDRS), e una meta-analisi ha mostrato che rAAV2/2-ND4 era più efficace dell’idebenone, entrambi più efficaci del decorso naturale. 2)
Attualmente non approvato da EMA e FDA, l’efficacia nella malattia di Harding non è stata verificata. Si prevede un effetto sulla componente LHON, mentre l’impatto sulla patologia della SM non è chiaro. 2)
È in fase di studio una tecnica che prevede la somministrazione intravitreale di MSC derivate da iPSC per trasferire direttamente i mitocondri alle CGR. I nanotubi di tunneling (TNT) dipendenti da F-actina mediano il trasferimento. È stata riportata la prevenzione della riduzione della densità delle CGR in topi Ndufs4 KO, ma non si è ancora arrivati a studi clinici. 3)
È stato suggerito un possibile miglioramento dei potenziali evocati visivi (PEV) nella SM, e si sta valutando l’applicazione in pazienti con malattia di Harding con prognosi visiva sfavorevole.
Sono necessari lo sviluppo di biomarcatori specifici per la malattia di Harding e studi prospettici. Il trattamento di questa malattia, che combina le patologie di LHON e SM, dovrebbe essere personalizzato, ed è indispensabile una collaborazione multidisciplinare.
QLa terapia genica può essere utilizzata anche per la malattia di Harding?
A
rAAV2/2-ND4 è una terapia genica per pazienti LHON con mutazione MT-ND4 e si prevede un effetto sulla componente LHON. 2)Tuttavia, l’efficacia per la malattia di Harding non è stata verificata e attualmente non è approvata da EMA e FDA. L’impatto sulla patologia della SM non è chiaro e per l’applicazione a questa malattia è necessaria un’attenta valutazione.
Alorainy J, Alorfi Y, Karanjia R, Badeeb N. A Comprehensive Review of Leber Hereditary Optic Neuropathy and Its Association with Multiple Sclerosis-Like Phenotypes Known as Harding’s Disease. Eye and brain. 2024;16:17-24. doi:10.2147/EB.S470184. PMID:39100385; PMCID:PMC11296356.
Wong DCS, Makam R, Yu-Wai-Man P. Advanced therapies for inherited optic neuropathies. Eye (London, England). 2026;40(2):177-184. doi:10.1038/s41433-025-04109-1. PMID:41318849; PMCID:PMC12830909.
Hu JL, Hsu CC, Hsiao YJ, et al. Leber’s hereditary optic neuropathy: Update on the novel genes and therapeutic options. J Chin Med Assoc. 2024;87:12-16.
Sanders FWB, Votruba M. Outcomes of idebenone therapy for Leber hereditary optic neuropathy in a cohort of patients from Wales. Eye (Lond). 2025 Sep 17;39(16):2952-2957. doi:10.1038/s41433-025-03993-x. PMID:40962867; PMCID:PMC12583466.
Copia il testo dell'articolo e incollalo nell'assistente IA che preferisci.
Articolo copiato negli appunti
Apri un assistente IA qui sotto e incolla il testo copiato nella chat.