A doença de Harding é uma condição na qual sintomas neurológicos desmielinizantes semelhantes à esclerose múltipla (EM) coexistem com um histórico de mutação no DNA mitocondrial (mtDNA) da neuropatia óptica hereditária de Leber (LHON). O nome deriva do relato de Harding et al. em 1992 de oito mulheres com neuropatia óptica bilateral e histórico familiar de LHON, seis das quais apresentavam sintomas neurológicos compatíveis com EM. 1)
Até o momento, um total de 88 casos foram relatados na literatura. 1) 70,4% (62 casos) dos pacientes são mulheres, com uma proporção homem:mulher de 2,38:1. 1) Isso contrasta com a LHON, que é mais comum em homens (93,1% homens), sugerindo que a predisposição feminina à EM pode estar envolvida. A idade média de início é de 30,5 anos. 1)
A prevalência de LHON no Japão é de cerca de 1/50.000 (estima-se um total de 4.000 a 5.000 pacientes no país), no Reino Unido de 1/31.000 e na Austrália de 1/68.000. 1)Em 2015, a LHON foi designada como doença rara no Japão e foram estabelecidos critérios diagnósticos. O número de novos casos é de aproximadamente 117 por ano, com 47% dos casos ocorrendo até os 30 anos de idade.
QQuão rara é a doença de Harding?
A
Apenas 88 casos foram relatados na literatura, sendo uma doença extremamente rara. 1)70,4% dos pacientes são mulheres e a idade média de início é de 30,5 anos. Como requer a coexistência de duas doenças, LHON e EM, é muito menos comum do que LHON ou EM isoladamente.
Diminuição da visão começando com visão turva: começa com visão turva indolor, piora gradualmente, levando a grave perda de visão e escotoma central.
Deficiência binocular: geralmente afeta ambos os olhos. O intervalo até o segundo olho ser afetado é maior que na LHON, com média de 1,66 anos. 1)
Múltiplos episódios de deficiência visual: enquanto a LHON geralmente apresenta apenas dois episódios, a doença de Harding causa múltiplos episódios de redução da visão. 1)
Ausência de dor ocular: ao contrário da neurite óptica na EM, não há dor ocular. 1) Este é um ponto importante para o diagnóstico diferencial.
Sintomas extraoculares: podem incluir tremor postural, neuropatia periférica, distúrbios motores, arritmia cardíaca, fraqueza muscular e doenças musculares.
Achados clínicos (achados confirmados pelo médico no exame)
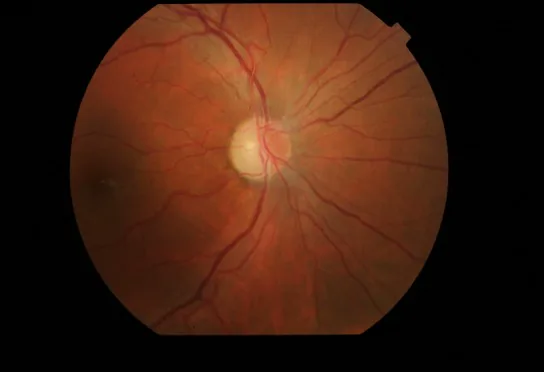
Hiperemia e rubor do disco óptico: acompanhado de edema da camada de fibras nervosas da retina (RNFL) ao redor do disco.
Telangiectasias retinianas: dilatação dos capilares (telangiectasia) e aumento do tortuosidade vascular.
Não vazamento na angiografia fluoresceínica: não se observa vazamento de corante fluorescente a partir do disco óptico hiperemiado. Este é um importante ponto de diferenciação da neurite óptica.
Achados de OCT: considera-se que há edema do disco óptico antes do início dos sintomas.
Fase crônica
Atrofia do disco óptico: a atrofia do disco óptico persiste após a resolução da inflamação.
Afinamento da RNFL: A OCT mostra afinamento das camadas internas da retina, principalmente do feixe papilomacular.
Redução da acuidade visual corrigida: Frequentemente permanece em torno de 0,01. A percepção luminosa é preservada.
Preservação do reflexo pupilar à luz: Comparado a outras neuropatias ópticas, o reflexo pupilar à luz é preservado ou apenas levemente prejudicado.
Campo visual: Apresenta escotoma central denso ou escotoma centrocecal.
QComo a perda de visão na doença de Harding difere da LHON e da neurite óptica na EM?
A
A diferença da LHON é que ocorrem múltiplos episódios de perda de visão e o intervalo até o segundo olho ser afetado é longo (média de 1,66 anos).1)A diferença da neurite óptica na EM é que não há dor ocular e não se observa extravasamento de corante da papila óptica na angiografia fluoresceínica. Essas características são pistas importantes para o diagnóstico diferencial da doença de Harding.
A causa da doença de Harding são mutações patogênicas no DNA mitocondrial (mtDNA). As três principais mutações codificam subunidades do complexo I, induzindo apoptose das células ganglionares da retina (RGC) por meio de síntese prejudicada de ATP e aumento de espécies reativas de oxigênio (ROS). 3)
A distribuição das mutações em 88 casos da doença de Harding é mostrada abaixo.
Mutação
Gene
Proporção na doença de Harding
m.11778G>A
MT-ND4
69,3% (61/88 casos)
m.14484T>C
MT-ND6
12,5%
m.3460G>A
MT-ND1
10,2%
Citação: Alorainy J et al. 20241)
A mutação MT-ND4 representa 90% dos casos de LHON na Ásia e 70% na Europa. 3) Em pacientes japoneses, essas três mutações genéticas representam 95% dos casos.
O mtDNA é transmitido de mãe para filho (herança materna). Não é transmitido aos descendentes de pacientes do sexo masculino.
Como mecanismo para a coexistência de LHON e EM, três hipóteses foram propostas. 1)
Teoria da modificação da LHON pela EM: a mutação do mtDNA modifica o fenótipo da EM, causando neurite óptica atípica indolor, mais grave e irreversível.
Teoria da manifestação da LHON por predisposição à EM: fatores genéticos e ambientais que predispõem à EM em mulheres promovem o início da LHON em portadores assintomáticos de mutação do mtDNA.
Teoria da indução inflamatória acidental: em pacientes com mutação do mtDNA da LHON, uma reação inflamatória acidental é induzida na via óptica anterior.
Tabagismo: pode estar envolvido como fator de risco para o desenvolvimento da LHON.
Consumo excessivo de álcool: também pode estar envolvido como fator de risco.
Medicamentos antituberculose (como etambutol): tem sido clinicamente apontado como fator envolvido.
QPor que a doença de Harding é mais comum em mulheres?
A
A LHON é predominante em homens (93,1% dos casos), enquanto na doença de Harding, 70,4% dos pacientes são mulheres. Isso está relacionado ao fato de a esclerose múltipla (EM) ser mais frequente em mulheres. Sugere-se que fatores genéticos e ambientais que predispõem à EM possam desencadear a LHON em portadores assintomáticos de mutações no mtDNA. 1) Além disso, a sinalização do estrogênio pode estar envolvida na patogênese da LHON. 2)
A doença de Harding é definida como “pacientes que preenchem os critérios diagnósticos de EM e possuem uma mutação primária do LHON” (Pfeffer et al.).
No Japão, os critérios diagnósticos para LHON estabelecidos em 2015 são aplicados.
Caso definitivo: preenche os sinais principais (perda visual aguda a subaguda, bilateral, indolor e escotoma central + um ou mais achados oftalmoscópicos anormais na fase aguda).
Caso definitivo: sinais principais + mutação missense no gene mitocondrial + ausência de anormalidade do nervo óptico retrobulbar na RM
Caso suspeito: herança materna evidente, mas mutação genética não detectável
Teste genético: No Japão, é possível solicitar externamente testes para as três mutações m.3460, m.11778 e m.14484. Se negativo, é necessário encaminhamento para uma instituição de referência.
Sequenciamento do exoma: Se a mutação do mtDNA não for identificada, detecta mutações em genes nucleares como DNAJC30.
Características de RM: Os achados que diferenciam a doença de Harding da EM incluem hipossinal e bordas mal definidas das lesões em T2, ausência de hipersinal em T1 e áreas de hipersinal ao redor dos cornos anteriores (diferente do padrão típico de dedos de Dawson). 1) 73% e 90% dos casos de LHON-EM preenchem os critérios de disseminação espacial de McDonald. 1)
Frequência crítica de fusão (FCF) e reflexo pupilar à luz: Preservados ou apenas levemente reduzidos.
Triagem: Em uma triagem de 1.666 pacientes com EM, apenas 5 apresentaram mutação positiva para LHON, não sendo recomendada a triagem de rotina. Recomenda-se avaliação caso a caso, limitada a pacientes com defeitos de campo visual graves ou bilaterais e/ou história familiar materna. 1)
Não há tratamento estabelecido. Abaixo estão as abordagens atuais.
Idebenona (não aprovado): Derivado da coenzima Q10. Auxilia na transferência de elétrons e pode manter ou melhorar a função visual. Ensaios clínicos foram realizados no Japão, com relatos de melhora visual em alguns casos, mas é um medicamento não aprovado no país. Alguns pacientes podem importá-lo para uso oral.
Suplementos: Coenzima Q10, vitaminas do complexo B e vitamina C são usados a critério de cada instituição.
Orientação para cessação do tabagismo: Como o tabagismo está associado ao risco de desenvolver LHON, recomenda-se parar de fumar.
Aconselhamento genético: Recomendado precocemente para mulheres que podem transmitir a mutação do mtDNA aos descendentes. Deve-se informar que a mutação não é transmitida aos descendentes de pacientes do sexo masculino, que há casos de recuperação espontânea e que a doença é reconhecida como doença rara.
Cuidados para baixa visão: fornecer cuidados adequados e orientação de vida para pacientes com deficiência visual residual.
Na ausência de um tratamento padrão estabelecido, os seguintes tratamentos estão sendo testados.
Metilprednisolona intravenosa: administração de 1g/dia por 3 dias, com relatos de melhora leve a moderada da visão em alguns pacientes. 1)
Mitoxantrona: Aproximadamente 19,2 mg/mês por via intravenosa mostrou melhora da visão e dos sintomas neurológicos, mas seu uso é limitado devido a efeitos colaterais graves. 1)
Plasmaférese e ciclofosfamida: Alguns relatos de melhora subjetiva na percepção de luz e sensibilidade ao contraste, mas sem consistência. 1)
Imunomoduladores: Proporcionam estabilidade clínica e de achados de ressonância magnética, mas não impedem a progressão do dano visual. Há relatos de rebote inflamatório após a interrupção do natalizumabe. 1)
QQue tratamento é realizado para a doença de Harding no Japão?
A
Não há tratamento estabelecido, sendo o tratamento sintomático o principal. Alguns pacientes importam pessoalmente a idebenona, não aprovada no país, para uso oral, mas as evidências de eficácia na doença de Harding são escassas.4) Atualmente, são realizados suplementos como CoQ10 e vitaminas do complexo B, orientação para cessação do tabagismo, cuidados de baixa visão e aconselhamento genético.
O mecanismo básico da LHON origina-se de uma deficiência na subunidade do complexo I (cadeia respiratória mitocondrial) devido a uma mutação no mtDNA. O comprometimento do transporte de elétrons reduz a síntese de ATP e, simultaneamente, acumula espécies reativas de oxigênio (ROS). Isso induz a apoptose das células ganglionares da retina (RGC), levando à atrofia do nervo óptico. 3)
A taxa de recuperação visual varia conforme a mutação: na mutação MT-ND4, a taxa de recuperação é de 4 a 25%, inferior à das mutações MT-ND1 e MT-ND6. 3) No Japão, a mutação mais comum, mt11778 (MT-ND4), apresenta uma taxa de melhora visual de apenas alguns por cento. A mutação mt14484 (MT-ND6) tem a maior taxa de melhora.
A maior penetrância da LHON em homens sugere o envolvimento de genes nucleares ligados ao cromossomo X. 3)
PRICKLE3 (cromossomo X, Xp11.23): regula a função da ATP sintase (complexo V). 3)
Mutação YARS2: prejudica a função dos complexos I, III e IV da cadeia de transporte de elétrons. 3)
Mutação DNAJC30 (c.152A>G): prejudica o mecanismo de reparo do complexo I, causando LHON de herança autossômica recessiva. 3)
Na histopatologia da LHON-MS, células T e macrófagos/micróglias ativados medeiam a lesão tecidual. A presença de células inflamatórias dentro das lesões da LHON é incomum, sugerindo um mecanismo imunológico inicial. 1) As alterações da substância branca contribuem não apenas com desmielinização semelhante à EM, mas também com vacuolização e palidez difusa da mielina. Disfunção mitocondrial, resposta autoimune e mimetismo molecular são mecanismos postulados para a desmielinização inflamatória. 1)
As sequelas neurológicas variam desde comprometimento leve até um curso semelhante à EM remitente-recorrente.
7. Pesquisas recentes e perspectivas futuras (relatos em fase de pesquisa)
Foi desenvolvida uma terapia genética (lenadogen nolparvoveque; rAAV2/2-ND4) para pacientes com LHON com mutação MT-ND4. A expressão gênica alotópica introduz a proteína ND4 do tipo selvagem nas mitocôndrias das CGR. 2)
No estudo REVERSE (2019), foi relatada uma melhora média na BCVA de -0,308 LogMAR no olho injetado intravítreo e -0,259 LogMAR no olho não tratado. 2) Esse resultado sugeriu uma transferência imunológica para o olho contralateral. A análise agrupada (ensaios RESCUE, REVERSE, RESTORE, REFLECT) mostrou uma melhora de aproximadamente 21,5 letras ETDRS em comparação com o grupo controle. A administração bilateral foi mais eficaz que a unilateral (cerca de 12 vs 8 letras ETDRS), e a meta-análise mostrou que rAAV2/2-ND4 foi mais eficaz que a idebenona, sendo ambos mais eficazes que a história natural. 2)
Atualmente não aprovado pela EMA e FDA, sua eficácia para a doença de Harding não foi verificada. Embora se espere um efeito no componente LHON, o impacto na patologia da EM não é claro. 2)
Está sendo estudada a administração intravítrea de MSC derivadas de iPSC para transferir diretamente mitocôndrias para as CGR. Os nanotúbulos de tunelamento (TNT) dependentes de F-actina medeiam a transferência. Em camundongos Ndufs4 KO, foi relatada a prevenção da redução da densidade de CGR, mas ainda não chegou a ensaios clínicos. 3)
Foi sugerida a possibilidade de melhora do potencial evocado visual (VEP) na EM, e sua aplicação está sendo considerada em pacientes com doença de Harding de mau prognóstico visual.
O desenvolvimento de biomarcadores específicos para a doença de Harding e estudos prospectivos são necessários. O tratamento desta doença, que apresenta patologia dupla de LHON e EM, deve ser individualizado, sendo essencial a colaboração multiprofissional.
QA terapia genética também pode ser usada para a doença de Harding?
A
rAAV2/2-ND4 é uma terapia genética para pacientes com LHON com mutação MT-ND4, e espera-se que tenha efeito no componente LHON. 2) No entanto, sua eficácia na doença de Harding não foi verificada e atualmente não é aprovada pela EMA e FDA. O impacto na patologia da EM também não está claro, e sua aplicação nesta doença requer consideração cuidadosa.
Alorainy J, Alorfi Y, Karanjia R, Badeeb N. A Comprehensive Review of Leber Hereditary Optic Neuropathy and Its Association with Multiple Sclerosis-Like Phenotypes Known as Harding’s Disease. Eye and brain. 2024;16:17-24. doi:10.2147/EB.S470184. PMID:39100385; PMCID:PMC11296356.
Wong DCS, Makam R, Yu-Wai-Man P. Advanced therapies for inherited optic neuropathies. Eye (London, England). 2026;40(2):177-184. doi:10.1038/s41433-025-04109-1. PMID:41318849; PMCID:PMC12830909.
Hu JL, Hsu CC, Hsiao YJ, et al. Leber’s hereditary optic neuropathy: Update on the novel genes and therapeutic options. J Chin Med Assoc. 2024;87:12-16.
Sanders FWB, Votruba M. Outcomes of idebenone therapy for Leber hereditary optic neuropathy in a cohort of patients from Wales. Eye (Lond). 2025 Sep 17;39(16):2952-2957. doi:10.1038/s41433-025-03993-x. PMID:40962867; PMCID:PMC12583466.
Copie o texto do artigo e cole no assistente de IA de sua preferência.
Artigo copiado para a área de transferência
Abra um assistente de IA abaixo e cole o texto copiado na conversa.