Doyne Honeycomb Retinal Distrofisi (DHRD), arka kutup ve optik disk çevresinde beyaz drusen benzeri birikintilerin radyal olarak dizildiği otozomal dominant kalıtımlı bir retina distrofisidir. Ayrıca “Malattia Leventinese (MLVT)” ve “familyal dominant drusen” olarak da adlandırılır. OMIM numarası #126600’dür. 1)
1899’da İngiliz göz doktoru Robert Doyne, dört kız kardeşte bu bulguyu gözlemlemiş ve ilk kez “petek deseni” olarak tanımlamıştır. 1925’te İsviçreli Vogt, Leventine Vadisi’nde aynı fenotipi rapor etmiş ve “Malattia Leventinese” adını vermiştir. 1999’da Stone ve arkadaşları, hem DHRD hem de MLVT ailelerinde EFEMP1 geninde aynı mutasyonu (R345W) tanımlayarak iki hastalığın aynı olduğunu kanıtlamıştır.
Sorumlu gen, kromozom 2p16.1’de yer alan ve fibulin 3 proteinini kodlayan EFEMP1’dir (EGF içeren fibulin benzeri hücre dışı matriks proteini 1). 2) Bu missense mutasyonu Arg345Trp (R345W), Bruch membranı ile retina pigment epiteli (RPE) arasında anormal birikintilere neden olarak drusen oluşumuna yol açar. 1)Yaşa bağlı makula dejenerasyonu ile drusen, Bruch membran kalınlaşması, RPE atrofisi ve kompleman aktivasyonu ortak patolojiyi paylaşır. 1)
QDHRD ve yaşa bağlı makula dejenerasyonu arasındaki fark nedir?
A
Yaşa bağlı makula dejenerasyonu, temel nedeni yaşlanma olan multifaktöriyel bir hastalıktır ve yaşlılarda ortaya çıkar. Buna karşılık DHRD, EFEMP1’deki R345W mutasyonu gibi tek bir gen mutasyonundan kaynaklanır, semptomlar genç-orta yaşta (40-50’li yaşlar) ortaya çıkar, aile öyküsü vardır ve otozomal dominant kalıtım gösterir. Drusen dağılımı, arka kutup ve optik disk çevresinde radyal dizilim ile karakteristiktir. Her ikisi de drusen oluşumu, Bruch membran değişiklikleri ve RPE atrofisi gibi ortak patofizyolojik yolları paylaştığından, DHRD yaşa bağlı makula dejenerasyonu için bir araştırma modeli olarak dikkat çekmektedir.
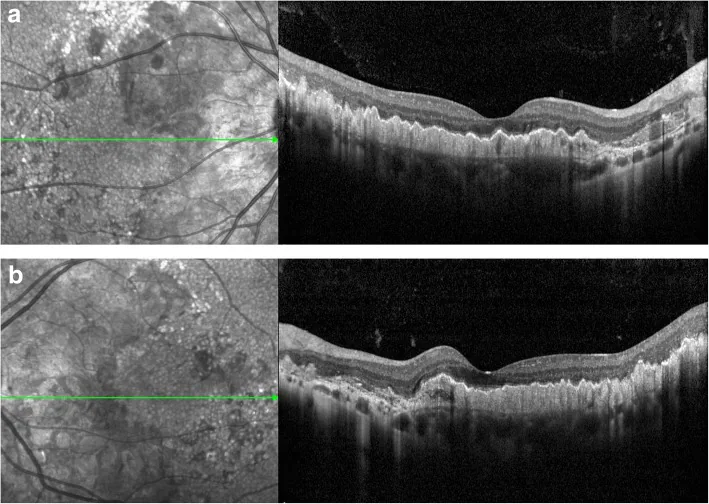
Arka kutup ve optik disk çevresinde drusen: Vasküler ark boyunca erken başlangıçlı drusen. Radyal düzen karakteristiktir.1)
İki tip drusen: Büyük yuvarlak drusenler ve küçük radyal drusenler olmak üzere iki tip görülür.2)
OKT bulguları: RPE/Bruch membran kompleksinde diffüz değişiklikler gösterir. Nörosensoriyel tabaka nispeten korunur. Drusen benzeri PED ve çift katman işareti doğrulanır.1)2)
İleri evre bulguları
Drusen füzyonu ve pigment değişiklikleri: Drusenler birleşir ve RPE’de pigment anormalliği oluşur.2)
RPE atrofisi ve skarlaşması: Santral coğrafi atrofiye ilerleyebilir.2)
CNVM komplikasyonu: Nadirdir ancak görme azalmasının ana nedenidir. Fibrovasküler PED görülebilir.2)
Elektroretinogram amplitüd düşüklüğü: Tam alan elektroretinogramda amplitüd düşüklüğü doğrulanır. 1)
Multimodal görüntüleme ile değerlendirme, tanı ve takipte faydalıdır. 2)Fundus otofloresansında (FAF), drusen bölgesinde yüksek otofloresans, RPE atrofisi bölgesinde düşük otofloresans görülür. 2) Flicker eşik testinde (FDT) görme alanı duyarlılığında azalma saptanır. 1)
QSemptomların şiddeti kişiden kişiye değişir mi?
A
Aynı aile içinde bile şiddet farklılık gösterebilir. Bazı vakalarda ileri evreye kadar iyi görme korunurken, CNVM ile komplike olan vakalarda ani görme kaybı meydana gelebilir. Düzenli fundus muayenesi ile CNVM’nin erken tespiti önemlidir.
DHRD, tek bir gen mutasyonuna bağlı otozomal dominant kalıtımlı bir hastalıktır. Genetik bilgiler aşağıda verilmiştir.
Genetik bilgilerin özeti aşağıda verilmiştir.
Öğe
İçerik
Neden olan gen
EFEMP1 (2p16.1)
Mutasyon
Ekson 10 · R345W
Kalıtım şekli
Otozomal dominant
Kodlanan protein
Fibulin 3
EFEMP1, EGF içeren fibrilin benzeri hücre dışı matriks proteini 1’i kodlar ve Bruch membranının hücre dışı matriks bileşeni olarak işlev görür. R345W mutasyonu, protein katlanmasında anormalliğe neden olur ve RPE ile Bruch membranı arasında bazal katman birikintileri birikir. 1) Ayrıca, EFEMP1 mutasyonunun EGFR sinyalini baskılayarak kolesterol atılımında rol oynayan CES1’i inhibe ettiği ve lipid birikimini teşvik ettiği düşünülmektedir. Yaşa bağlı makula dejenerasyonu ve DHRD fare modellerinde kompleman aktivasyonunda artış da gözlenir. 1)
DHRD tanısı, genç yaşta başlangıç, aile öyküsü ve karakteristik drusen dağılım paterninin tanınmasıyla başlar. 2) Multimodal görüntüleme ile değerlendirme ve genetik testin kombinasyonu kesin tanı için faydalıdır. 2)
Örüntü tanıma: Genç yaşta (40-50’li yaşlar) başlangıç, iki taraflı simetrik tutulum, arka kutup ve optik disk çevresinde radyal drusen dağılımı tanısal ipuçlarıdır. 2)
OCT: RPE/Bruch membran kompleks değişiklikleri, drusen benzeri PED, çift katman işareti ve nörosensöriyel tabakanın korunmasını doğrulayın. 2)
Fundus otofloresans (FAF): Drusenlerde yüksek otofloresans ve RPE atrofisi alanlarında düşük otofloresans paternini değerlendirin. 2)
Genetik test: NextSeq 550 (Illumina) ile yeni nesil dizileme kullanarak EFEMP1 mutasyonunu doğrulayın (≥20× kapsama). ACMG kılavuzuna göre c.1033C>T (R345W) patojenik mutasyon olarak sınıflandırılır (PM2, PP3, PP5 kriterleri). 1)
QKesin tanı için genetik test mutlaka gerekli midir?
A
Klinik bulgular tipik olduğunda (genç yaşta başlangıç, iki taraflı simetrik arka kutup druseni, aile öyküsü) genetik test olmadan klinik tanı mümkündür. Ancak kesin tanı, aile taraması ve genetik danışmanlık amacıyla EFEMP1’de R345W mutasyonunun doğrulanması önerilir.
Anti-VEGF intravitreal enjeksiyon, CNVM ile birlikte görülen olgularda bir tedavi seçeneğidir. Ranibizumab (0.5 mg) intravitreal enjeksiyonu kullanılır ve enjeksiyon sonrası görme keskinliğinde iyileşme ve seröz retina dekolmanının (SRF) kaybolduğu bildirilmiştir. 2)
Parameswarappa ve Rani, tip 1 koroidal neovasküler membranı olan 44 yaşında bir kadın (DHRD) bildirdi; bir kez ranibizumab enjeksiyonu sonrası BCVA 20/40’tan 20/30’a iyileşti ve SRF kayboldu. 2) CNVM tedavisi için fotodinamik tedavi (PDT·verteporfin) raporları da mevcuttur. 2)
QNanosecond lazer (2RT) tedavisi alınabilir mi?
A
2RT (nanosaniye puls lazeri), DHRD için yeni bir tedavi olarak vaka raporlarında fonksiyonel iyileşme göstermiştir, ancak şu anda araştırma aşamasındadır ve standart tedavi olarak yerleşmemiştir. Ayrıntılar için “En Son Araştırmalar ve Gelecek Perspektifleri” bölümüne bakın.
EFEMP1 (EGF içeren fibulin benzeri hücre dışı matriks protein 1), Bruch membranının hücre dışı matriks bileşeni olarak işlev görür.1) R345W mutasyonu, proteinin anormal katlanmasına neden olur ve RPE ile Bruch membranı arasında bazal katman birikintileri birikir.1) Bu birikintiler drusen oluşumunun temelini oluşturur.
Drusen oluşumu: Bruch membranında anormal madde birikimi
Bruch membran kalınlaşması: Membranın yapısal değişikliklerine bağlı geçirgenlik azalması
RPE atrofisi: Beslenme bozukluğuna bağlı pigment epitel dejenerasyonu
Kompleman aktivasyonu: Her iki hastalığın fare modellerinde kompleman sisteminin aşırı aktivasyonu doğrulanmıştır
EGFR sinyali üzerindeki etki olarak, EFEMP1 R345W mutasyonu EGFR yolunu aşırı baskılar ve kolesterol atılımında rol oynayan CES1 ekspresyonunu azaltır. Bu, lipid birikimini hızlandırarak drusen oluşumuna yol açar.
Elektrofizyolojik incelemelerde, tam alan elektroretinografide amplitüd düşüklüğü gözlenir ve bu, hem çubuk hem de koni hücrelerinin işlev bozukluğuna işaret eder.1)FDT görme alanı testinde, görme alanı duyarlılığında anlamlı bir azalma doğrulanmıştır.1)
QDHRD, yaşa bağlı makula dejenerasyonu araştırmalarına neden yardımcı olur?
A
DHRD, yaşa bağlı makula dejenerasyonu ile aynı moleküler ve patolojik yolları (Bruch membran değişiklikleri, RPE atrofisi, kompleman aktivasyonu) paylaşmasına rağmen, tek bir gen mutasyonu (R345W) ile ortaya çıktığı için patolojideki nedensel ilişkiler net bir şekilde analiz edilebilir. Yaşa bağlı makula dejenerasyonu, genomik ve çevresel çok faktörlü bir hastalıktır ve mekanizma analizi zordur; ancak DHRD modeli, ortak mekanizmaları araştırmak için uygun bir sistem sağlar.
7. Güncel Araştırmalar ve Gelecek Perspektifleri (Araştırma Aşamasındaki Raporlar)
2RT (2 Dakikalık Retina Tedavisi), ultra düşük enerjili nanosaniye darbe lazeri (400 μm çap, 3 nanosaniye, 532 nm, 0.15-0.45 mJ) kullanan non-invaziv bir tedavi yöntemidir. RPE debridmanı ve yara iyileşme yanıtı yoluyla katman yeniden oluşumunu teşvik ettiği varsayılan bir mekanizma öngörülmektedir. 1)
Cusumano ve ark. (2023), DHRD’li 3 hastada (41-46 yaş) 2RT tedavisi uygulamış ve en uzun 30 aylık takibi rapor etmiştir. 1) Başlıca sonuçlar şöyledir:
Görme keskinliği: Olgu 1’de 2-10 harf iyileşme gözlendi
FDT görme alanı duyarlılığı: Olgu 2’de sağ göz MD −12 dB’den, Olgu 3’te sol göz MD −9 dB’den iyileşme gözlendi
Tam alan elektroretinogram amplitüdü: Olgu 1 ve 2’de anlamlı artış doğrulandı
Güvenlik: Tedaviyle ilişkili advers olay gözlenmedi (ancak Olgu 1’de 24. ayda kistoid makula ödemi ortaya çıktı)
Ayrıca, çubuk hücrelerine özgü elektroretinogram iyileşmesi (tam alan ERG’de iyileşme, multifokal elektroretinogramda değişiklik yok) gözlenmiş olup, başlıca çubuk sistemini hedef alan bir etki mekanizması düşündürmektedir. 1) Ayrıca tedavi edilmeyen diğer gözde de fonksiyonel iyileşme görülen vakalar olmuş ve sistemik dolaylı indüksiyon etkisi olasılığı tartışılmaktadır. 1)
Bu rapor, yalnızca bir vaka serisi (3 vaka) düzeyinde kanıt sunmaktadır ve büyük ölçekli randomize kontrollü çalışma yapılmamıştır. Tekrarlanan lazer seanslarının etkinliği ve optimal protokolün belirlenmesi gelecekteki zorluklar olarak belirtilmiştir. 1)
Cusumano A, Falsini B, D’Ambrosio M, et al. Long-term structural and functional assessment of Doyne honeycomb retinal dystrophy following nanosecond 2RT laser treatment: a case series. Case Rep Ophthalmol. 2023;14:626-639.
Parameswarappa DC, Rani PK. Utility of pattern recognition and multimodal imaging in the diagnosis and management of doyne honeycomb retinal dystrophy complicated with type one choroidal neovascular membrane. BMJ Case Rep. 2021;14:e237635.
Tsang SH, Sharma T. Doyne Honeycomb Retinal Dystrophy (Malattia Leventinese, Autosomal Dominant Drusen). Adv Exp Med Biol. 2018;1085:97-102. PMID: 30578491.
Makale metnini kopyalayıp tercih ettiğiniz yapay zeka asistanına yapıştırabilirsiniz.
Makale panoya kopyalandı
Aşağıdaki yapay zeka asistanlarından birini açın ve kopyalanan metni sohbet kutusuna yapıştırın.