La distrofia retinica a nido d’ape di Doyne (Doyne Honeycomb Retinal Dystrophy; DHRD) è una distrofia retinica autosomica dominante caratterizzata da depositi biancastri simili a drusen disposti radialmente nella regione posteriore e peripapillare. È anche nota come Malattia Leventinese (MLVT) o drusen familiare dominante. Il numero OMIM è #126600. 1)
Nel 1899, l’oculista britannico Robert Doyne osservò questa condizione in quattro sorelle e la descrisse per la prima volta come un “pattern a nido d’ape”. Nel 1925, lo svizzero Vogt riportò lo stesso fenotipo nella valle di Leventine, denominandolo “Malattia Leventinese”. Nel 1999, Stone e colleghi identificarono la stessa mutazione (R345W) nel gene EFEMP1 in famiglie con DHRD e MLVT, dimostrando che si tratta della stessa malattia.
Il gene responsabile è EFEMP1 (EGF-containing fibulin-like extracellular matrix protein 1), situato sul cromosoma 2p16.1, che codifica per la proteina fibulina 3. 2) La mutazione missenso Arg345Trp (R345W) causa depositi anomali tra la membrana di Bruch e l’epitelio pigmentato retinico (RPE), portando alla formazione di drusen. 1) La degenerazione maculare legata all’età e la distrofia retinica a nido d’ape condividono patologie comuni come drusen, ispessimento della membrana di Bruch, atrofia dell’RPE e attivazione del complemento. 1)
QIn che cosa differiscono la DHRD e la degenerazione maculare legata all'età?
A
La degenerazione maculare legata all’età è una malattia multifattoriale causata principalmente dall’invecchiamento e si manifesta negli anziani. La DHRD, invece, è causata da una mutazione genetica singola, la R345W del gene EFEMP1, con sintomi che compaiono in età giovane o media (40-50 anni) e segue una trasmissione autosomica dominante con storia familiare. La distribuzione dei drusen è caratteristica: disposizione radiale attorno al polo posteriore e alla papilla ottica. Poiché entrambe condividono un percorso patologico comune di formazione di drusen, alterazioni della membrana di Bruch e atrofia dell’RPE, la DHRD è considerata un modello di studio per la degenerazione maculare legata all’età.
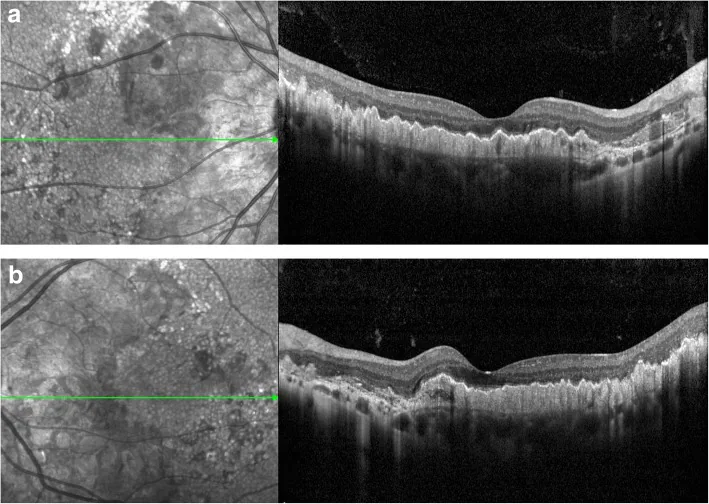
Kaiyan Zhang; Xuyang Sun; Yingying Chen; Qionglei Zhong; Lin Lin; Yuan Gao; Fanlin Hong. Doyne honeycomb retinal dystrophy/malattia leventinese induced by EFEMP1 mutation in a Chinese family. BMC Ophthalmol. 2018 Dec 12; 18:318. Figure 3. PMCID: PMC6292057. License: CC BY.
Scansione OCT. È stato notato un ispessimento iperriflettente sotto l’RPE accompagnato da un sollevamento ondulato (a: occhio destro; b: occhio sinistro).
Scotoma: si presenta come scotoma centrale o paracentrale.
Discromatopsia: osservata negli stadi avanzati.
Riduzione dell’acuità visiva: in caso di complicanza da CNVM, si verifica una marcata riduzione dell’acuità visiva. Nelle fasi avanzate, l’acuità visiva può scendere a 20/200 o meno. 2)
Drusen del polo posteriore e peripapillari: drusen a esordio precoce lungo l’arco vascolare. Caratteristica disposizione radiale. 1)
Due tipi morfologici di drusen: si osservano due tipi: grandi drusen rotondi e piccoli drusen radiali. 2)
Reperti OCT: mostrano alterazioni diffuse del complesso RPE/membrana di Bruch. Lo strato neurosensoriale è relativamente conservato. Si osservano PED simil-drusen e segno del doppio strato. 1)2)
Segni di stadio avanzato
Fusione di drusen e alterazioni pigmentarie: i drusen si fondono e si verificano anomalie pigmentarie dell’RPE. 2)
Atrofia e cicatrizzazione dell’RPE: può progredire verso un’atrofia geografica centrale. 2)
Complicanza da CNVM: rara ma principale causa di riduzione dell’acuità visiva. Può presentarsi come PED fibrovascolare. 2)
Riduzione dell’ampiezza dell’elettroretinogramma: l’elettroretinogramma a campo totale mostra una riduzione dell’ampiezza. 1)
La valutazione mediante imaging multimodale è utile per la diagnosi e il follow-up. 2) All’autofluorescenza del fondo oculare (FAF), si osserva iperautofluorescenza nelle aree di drusen e ipoautofluorescenza nelle aree di atrofia dell’RPE. 2) Il test di soglia di flicker (FDT) rileva una riduzione della sensibilità del campo visivo. 1)
QLa gravità dei sintomi varia da persona a persona?
A
Anche all’interno della stessa famiglia, la gravità può variare. In alcuni casi, una buona acuità visiva si mantiene fino a stadi avanzati, mentre nei casi complicati da CNVM si può verificare un improvviso calo visivo. È importante la diagnosi precoce della CNVM mediante esami periodici del fondo oculare.
La DHRD è una malattia autosomica dominante causata da una mutazione in un singolo gene. Di seguito sono riportate le informazioni genetiche.
Di seguito è riportata una panoramica delle informazioni genetiche.
Voce
Contenuto
Gene causativo
EFEMP1 (2p16.1)
Mutazione
Esone 10 · R345W
Modalità di trasmissione
Autosomica dominante
Proteina codificata
fibulina 3
EFEMP1 codifica per la proteina 1 della matrice extracellulare simile alla fibrillina contenente EGF e funge da componente della matrice extracellulare della membrana di Bruch. La mutazione R345W causa un ripiegamento anomalo della proteina, portando all’accumulo di depositi nello strato basale tra l’RPE e la membrana di Bruch. 1) Inoltre, si ritiene che la mutazione EFEMP1 inibisca CES1, coinvolta nell’efflusso di colesterolo, attraverso la soppressione del segnale EGFR, promuovendo l’accumulo di lipidi. Nei modelli murini di degenerazione maculare legata all’età e DHRD si osserva anche un’aumentata attivazione del complemento. 1)
La diagnosi di DHRD inizia con il riconoscimento dell’esordio giovanile, della storia familiare e del pattern caratteristico di distribuzione delle drusen. 2) La combinazione di valutazione mediante imaging multimodale e test genetici è utile per la diagnosi definitiva. 2)
Riconoscimento di pattern: esordio giovanile (40-50 anni), bilateralità simmetrica, distribuzione radiale di drusen al polo posteriore e intorno alla papilla sono indizi diagnostici. 2)
OCT: conferma di alterazioni del complesso RPE/membrana di Bruch, PED simile a drusen, segno del doppio strato e mantenimento dello strato neurosensoriale. 2)
Autofluorescenza del fondo (FAF): valutazione del pattern di iperautofluorescenza dei drusen e ipoautofluorescenza delle aree di atrofia dell’RPE. 2)
Test genetico: sequenziamento di nuova generazione con NextSeq 550 (Illumina) per confermare la mutazione EFEMP1 (copertura ≥20×). Secondo le linee guida ACMG, c.1033C>T (R345W) è classificata come mutazione patogenica (criteri PM2, PP3, PP5). 1)
QIl test genetico è sempre necessario per la diagnosi definitiva?
A
Se i reperti clinici sono tipici (esordio giovanile, drusen simmetrici bilaterali del polo posteriore, anamnesi familiare), la diagnosi clinica è possibile anche senza test genetico. Tuttavia, ai fini della diagnosi definitiva, dello screening familiare e della consulenza genetica, si raccomanda la conferma della mutazione R345W di EFEMP1.
Attualmente non esiste una terapia modificante la malattia consolidata per la DHRD. La strategia terapeutica varia a seconda della presenza o meno di CNVM.
Iniezione intravitreale di anti-VEGF è un’opzione terapeutica per i casi con CNVM. Viene utilizzata l’iniezione intravitreale di ranibizumab (0,5 mg) e dopo l’iniezione sono stati riportati miglioramento dell’acuità visiva e risoluzione del distacco sieroso retinico (SRF). 2)
Parameswarappa e Rani hanno riportato il caso di una donna di 44 anni (DHRD) con membrana neovascolare coroidale di tipo 1, che dopo una singola iniezione di ranibizumab ha mostrato un miglioramento della BCVA da 20/40 a 20/30 e risoluzione del SRF. 2) Esistono anche segnalazioni di trattamento della CNVM con terapia fotodinamica (PDT con verteporfina). 2)
QÈ possibile ricevere il trattamento con laser a nanosecondi (2RT)?
A
Il 2RT (laser a impulsi nanosecondi) è stato riportato in case report come nuovo trattamento per la DHRD con miglioramenti funzionali, ma al momento è ancora in fase di ricerca e non è stabilito come trattamento standard. Per maggiori dettagli, si veda la sezione “Ultime ricerche e prospettive future”.
6. Fisiopatologia e meccanismo dettagliato di insorgenza
EFEMP1 (proteina 1 della matrice extracellulare simile alla fibulina contenente EGF) funge da componente della matrice extracellulare della membrana di Bruch. 1) La mutazione R345W causa un ripiegamento anomalo della proteina, portando all’accumulo di depositi nello strato basale tra l’RPE e la membrana di Bruch. 1) Questi depositi costituiscono la base per la formazione di drusen.
La DHRD e la degenerazione maculare legata all’età condividono i seguenti percorsi patologici. 1)
Formazione di drusen: deposizione di materiale anomalo sulla membrana di Bruch
Ispessimento della membrana di Bruch: ridotta permeabilità dovuta a cambiamenti strutturali della membrana
Atrofia dell’RPE: degenerazione dell’epitelio pigmentato per alterazioni nutrizionali
Attivazione del complemento: in modelli murini di entrambe le malattie è stata confermata un’iperattivazione del sistema del complemento
Per quanto riguarda l’effetto sulla segnalazione dell’EGFR, la mutazione EFEMP1 R345W sopprime eccessivamente la via dell’EGFR, riducendo l’espressione di CES1, coinvolta nell’escrezione del colesterolo. Ciò promuove l’accumulo di lipidi e si ritiene porti alla formazione di drusen.
Studi elettrofisiologici hanno mostrato una riduzione dell’ampiezza dell’elettroretinogramma a campo totale, suggerendo una disfunzione sia dei coni che dei bastoncelli. 1) Il test del campo visivo FDT ha evidenziato una significativa riduzione della sensibilità del campo visivo. 1)
QPerché la DHRD è utile per la ricerca sulla degenerazione maculare legata all'età?
A
La DHRD condivide gli stessi percorsi molecolari e patologici della degenerazione maculare legata all’età (alterazioni della membrana di Bruch, atrofia dell’RPE, attivazione del complemento), ma essendo causata da una singola mutazione genetica (R345W), consente di analizzare chiaramente le relazioni causali della patologia. Mentre la degenerazione maculare legata all’età è una malattia multifattoriale con fattori genomici e ambientali, rendendo difficile l’analisi dei meccanismi, il modello DHRD rappresenta un sistema adatto per studiare i meccanismi comuni.
7. Ricerche recenti e prospettive future (rapporti in fase di studio)
La 2RT (2-minute Retina Treatment) è un trattamento non invasivo che utilizza un laser a impulsi nanosecondi a energia ultra-bassa (diametro 400 μm, 3 nanosecondi, 532 nm, 0,15-0,45 mJ). Si ipotizza che il meccanismo promuova la rimozione dell’RPE e la ristrutturazione degli strati attraverso la risposta di guarigione della ferita. 1)
Cusumano et al. (2023) hanno trattato 3 pazienti con DHRD (età 41-46 anni) con terapia 2RT, riportando un follow-up fino a 30 mesi. 1) I principali risultati sono stati i seguenti:
Acuità visiva: nel Caso 1 è stato osservato un miglioramento di 2-10 lettere
Sensibilità del campo visivo FDT: nel Caso 2 è stato osservato un miglioramento da OD MD −12 dB, nel Caso 3 da OS MD −9 dB
Ampiezza dell’elettroretinogramma a campo totale: è stato confermato un aumento significativo nei Casi 1 e 2
Sicurezza: non sono stati osservati eventi avversi correlati al trattamento (tuttavia, al mese 24 nel Caso 1 è comparso edema maculare cistoide)
Inoltre, è stato osservato un miglioramento dell’elettroretinogramma specifico dei bastoncelli (miglioramento dell’ERG a campo totale, nessuna variazione dell’ERG multifocale), suggerendo un meccanismo d’azione mirato principalmente al sistema dei bastoncelli. 1) Inoltre, in alcuni casi si è osservato un miglioramento funzionale anche nell’occhio controlaterale non trattato, suggerendo un possibile effetto di induzione indiretta sistemica. 1)
Questo rapporto si limita al livello di evidenza di una serie di casi (3 casi) e non è stato condotto alcun RCT su larga scala. L’efficacia delle sessioni laser ripetute e la definizione di un protocollo ottimale sono considerate sfide future. 1)
Cusumano A, Falsini B, D’Ambrosio M, et al. Long-term structural and functional assessment of Doyne honeycomb retinal dystrophy following nanosecond 2RT laser treatment: a case series. Case Rep Ophthalmol. 2023;14:626-639.
Parameswarappa DC, Rani PK. Utility of pattern recognition and multimodal imaging in the diagnosis and management of doyne honeycomb retinal dystrophy complicated with type one choroidal neovascular membrane. BMJ Case Rep. 2021;14:e237635.
Tsang SH, Sharma T. Doyne Honeycomb Retinal Dystrophy (Malattia Leventinese, Autosomal Dominant Drusen). Adv Exp Med Biol. 2018;1085:97-102. PMID: 30578491.
Copia il testo dell'articolo e incollalo nell'assistente IA che preferisci.
Articolo copiato negli appunti
Apri un assistente IA qui sotto e incolla il testo copiato nella chat.