La distrofia retiniana en panal de Doyne (DHRD, por sus siglas en inglés) es una distrofia retiniana autosómica dominante caracterizada por depósitos blancos similares a drusas dispuestos radialmente en el polo posterior y alrededor del disco óptico. También se conoce como Malattia Leventinese (MLVT) y drusas dominantes familiares. El número OMIM es #126600. 1)
En 1899, el oftalmólogo británico Robert Doyne observó este hallazgo en cuatro hermanas y lo describió por primera vez como un “patrón de panal”. En 1925, Vogt, de Suiza, informó el mismo fenotipo en el valle de Leventine y lo denominó “Malattia Leventinese”. En 1999, Stone y colaboradores identificaron la misma mutación (R345W) en el gen EFEMP1 en familias con DHRD y MLVT, demostrando que son la misma enfermedad.
El gen causante es EFEMP1 (proteína 1 de la matriz extracelular similar a fibulina que contiene EGF) ubicado en el cromosoma 2p16.1, que codifica la proteína fibulina 3. 2) Esta mutación sin sentido Arg345Trp (R345W) provoca depósitos anormales entre la membrana de Bruch y el epitelio pigmentario de la retina (EPR), lo que lleva a la formación de drusas. 1) Comparte vías patológicas comunes con la degeneración macular asociada a la edad, como drusas, engrosamiento de la membrana de Bruch, atrofia del EPR y activación del complemento. 1)
Q¿En qué se diferencia la DHRD de la degeneración macular asociada a la edad?
A
La degeneración macular asociada a la edad es una enfermedad multifactorial causada principalmente por el envejecimiento y se presenta en adultos mayores. En cambio, la DHRD es causada por una mutación genética única (R345W en EFEMP1), con síntomas que aparecen en adultos jóvenes a medianos (40-50 años), con antecedentes familiares y herencia autosómica dominante. La distribución de las drusas es característica, con disposición radial en el polo posterior y alrededor del disco óptico. Debido a que ambas comparten vías patológicas comunes de formación de drusas, cambios en la membrana de Bruch y atrofia del EPR, la DHRD también se considera un modelo de investigación para la degeneración macular asociada a la edad.
En las etapas iniciales, a menudo transcurre asintomático. Los siguientes síntomas aparecen entre los 40 y 50 años.
Visión borrosa y visión central borrosa: Puede acompañarse de disminución de la percepción de contraste. 1)
Metamorfopsia: Se percibe como distorsión en el área central.
Escotoma: Aparece como un escotoma central o paracentral.
Anomalía de la visión cromática: Se observa en la etapa avanzada.
Pérdida de agudeza visual: Cuando se asocia CNVM, se produce una pérdida marcada de la agudeza visual. En la etapa avanzada, la agudeza visual puede ser de 20/200 o peor. 2)
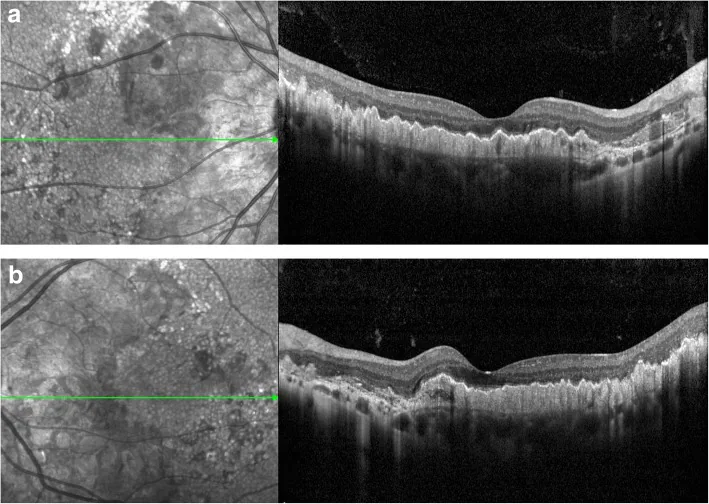
Drusas en el polo posterior y alrededor del disco óptico: Drusas de inicio temprano a lo largo de los arcos vasculares. La disposición radial es característica. 1)
Dos tipos de morfología de drusas: Se observan dos tipos: drusas redondas grandes y drusas radiales pequeñas. 2)
Hallazgos de OCT: Muestran cambios difusos en el complejo EPR/membrana de Bruch. La capa neurosensorial está relativamente preservada. Se confirman PED drusenoide y signo de doble capa. 1)2)
Hallazgos en etapa avanzada
Fusión de drusas y cambios pigmentarios: Las drusas se fusionan y se producen anomalías pigmentarias en el EPR. 2)
Atrofia y cicatrización del EPR: Puede progresar a atrofia geográfica central. 2)
Complicación de CNVM: Rara pero es una causa principal de pérdida de visión. Puede presentarse con PED fibrovascular. 2)
Amplitud reducida del electrorretinograma: Se confirma una amplitud reducida en el electrorretinograma de campo completo. 1)
La evaluación mediante imágenes multimodales es útil para el diagnóstico y el seguimiento. 2) La autofluorescencia del fondo de ojo (FAF) muestra hiperautofluorescencia en las áreas de drusas e hipoautofluorescencia en las áreas de atrofia del EPR. 2) La prueba de umbral de parpadeo (FDT) detecta una disminución de la sensibilidad del campo visual. 1)
Q¿Existe variación individual en la gravedad de los síntomas?
A
Incluso dentro de la misma familia, hay diferencias en la gravedad. En algunos casos, se mantiene una buena agudeza visual hasta etapas avanzadas, mientras que en casos complicados con CNVM, puede ocurrir una pérdida rápida de la visión. Los exámenes regulares del fondo de ojo son importantes para la detección temprana de CNVM.
La DHRD es un trastorno autosómico dominante causado por una mutación en un solo gen. La información genética se muestra a continuación.
A continuación se presenta un resumen de la información genética.
Elemento
Contenido
Gen causante
EFEMP1 (2p16.1)
Mutación
Exón 10, R345W
Patrón de herencia
Autosómico dominante
Proteína codificada
Fibulina 3
EFEMP1 codifica la proteína 1 de la matriz extracelular similar a fibulina que contiene EGF, que funciona como un componente de la matriz extracelular de la membrana de Bruch. La mutación R345W causa un plegamiento anormal de la proteína, lo que lleva a la acumulación de depósitos en la capa basal entre el EPR y la membrana de Bruch. 1) Además, se cree que la mutación de EFEMP1 inhibe CES1, involucrado en la salida de colesterol a través de la supresión de la señalización de EGFR, promoviendo así la acumulación de lípidos. También se observa una mayor activación del complemento en modelos de ratón de degeneración macular relacionada con la edad y DHRD. 1)
El diagnóstico de la DHRD comienza con el reconocimiento de la aparición temprana, los antecedentes familiares y los patrones característicos de distribución de drusas. 2) La combinación de evaluación por imágenes multimodales y pruebas genéticas es útil para el diagnóstico definitivo. 2)
Reconocimiento de patrones: La aparición en edad joven (40-50 años), bilateral simétrica y la distribución radial de drusas alrededor del polo posterior y el disco óptico son claves diagnósticas. 2)
OCT: Evaluar cambios en el complejo EPR/membrana de Bruch, PED drusenoide, signo de doble capa y preservación de la capa neurosensorial. 2)
Autofluorescencia de fondo (FAF): Evaluar la hiperautofluorescencia de las drusas y el patrón de hipoautofluorescencia en áreas de atrofia del EPR. 2)
Pruebas genéticas: Confirmar la mutación EFEMP1 mediante secuenciación de nueva generación con NextSeq 550 (Illumina) con cobertura ≥20×. Según las guías ACMG, c.1033C>T (R345W) se clasifica como variante patogénica (criterios PM2, PP3, PP5). 1)
Es importante diferenciar de enfermedades que muestran hallazgos similares en el fondo de ojo.
Enfermedad
Características
Puntos clave para la diferenciación
Degeneración macular relacionada con la edad
Inicio tardío, multifactorial
Edad de inicio, sin antecedentes familiares
Distrofia de Sorsby
Mutación TIMP3
Drusas reticulares
Enfermedad de Stargardt
Mutación ABCA4
Manchas maculares
MPGN tipo II
Enfermedad sistémica
Con disfunción renal
Q¿Es siempre necesaria la prueba genética para un diagnóstico definitivo?
A
Si los hallazgos clínicos son típicos (inicio juvenil, drusas del polo posterior bilaterales simétricas, antecedentes familiares), es posible un diagnóstico clínico sin pruebas genéticas. Sin embargo, para el diagnóstico definitivo, el cribado familiar y el asesoramiento genético, se recomienda la confirmación de la mutación R345W en EFEMP1.
Actualmente no existe una terapia modificadora de la enfermedad establecida para la DHRD. La estrategia de tratamiento difiere según la presencia o ausencia de CNVM.
Seguimiento regular: Se realiza examen de fondo de ojo y OCT para la detección temprana de CNVM.
Rehabilitación visual: Si la agudeza visual está significativamente reducida en la etapa avanzada, se proporciona rehabilitación visual y entrenamiento de movilidad. 2)
La inyección intravítrea de anti-VEGF es una opción de tratamiento para los casos con CNVM. Se utiliza la inyección intravítrea de ranibizumab (0.5 mg), y se ha informado mejoría de la agudeza visual y resolución del desprendimiento seroso de retina (SRF) después de la inyección. 2)
Parameswarappa y Rani informaron de una mujer de 44 años (DHRD) que desarrolló una membrana neovascular coroidea tipo 1, y tras una sola inyección de ranibizumab, la AVCC mejoró de 20/40 a 20/30, con resolución del LSR. 2) También hay informes de tratamiento de CNVM con terapia fotodinámica (PDT/verteporfina). 2)
Q¿Se puede recibir tratamiento con láser de nanosegundos (2RT)?
A
El 2RT (láser de pulso de nanosegundos) ha sido reportado en casos clínicos como un nuevo tratamiento para DHRD que mejora la función, pero actualmente es un tratamiento en investigación y no está establecido como terapia estándar. Para más detalles, consulte la sección “Investigación más reciente y perspectivas futuras”.
EFEMP1 (proteína 1 de la matriz extracelular similar a fibulina que contiene EGF) funciona como un componente de la matriz extracelular de la membrana de Bruch. 1) La mutación R345W causa un plegamiento anormal de la proteína, lo que lleva a la acumulación de depósitos de la capa basal entre el EPR y la membrana de Bruch. 1) Estos depósitos son la base de la formación de drusas.
Formación de drusas: depósito anormal de material en la membrana de Bruch
Engrosamiento de la membrana de Bruch: disminución de la permeabilidad debido a cambios estructurales
Atrofia del EPR: degeneración del epitelio pigmentario por deficiencia nutricional
Activación del complemento: se ha confirmado la activación del sistema del complemento en modelos murinos de ambas enfermedades
En cuanto al impacto en la señalización de EGFR, la mutación EFEMP1 R345W suprime en exceso la vía de EGFR y reduce la expresión de CES1, involucrada en la salida de colesterol. Esto promueve la acumulación de lípidos y se cree que conduce a la formación de drusas.
Los estudios electrofisiológicos muestran una amplitud reducida en el electrorretinograma de campo completo, lo que sugiere disfunción tanto de bastones como de conos. 1) La perimetría FDT confirma una disminución significativa de la sensibilidad del campo visual. 1)
Q¿Por qué es útil la DHRD para la investigación de la degeneración macular asociada a la edad?
A
La DHRD comparte las mismas vías moleculares y patológicas que la degeneración macular asociada a la edad (cambios en la membrana de Bruch, atrofia del EPR, activación del complemento), pero es causada por una mutación genética única (R345W), lo que permite un análisis claro de las relaciones causales. Mientras que la degeneración macular asociada a la edad es una enfermedad multifactorial que involucra factores genómicos y ambientales, lo que dificulta el análisis mecanicista, el modelo DHRD proporciona un sistema adecuado para estudiar estos mecanismos comunes.
7. Últimas investigaciones y perspectivas futuras (informes en etapa de investigación)
2RT (Tratamiento de Retina en 2 minutos) es un tratamiento no invasivo que utiliza un láser de pulso de nanosegundos de ultra baja energía (diámetro 400μm, 3 nanosegundos, 532nm, 0.15-0.45mJ). Se hipotetiza que promueve el desbridamiento del EPR y la reformación de capas a través de la respuesta de cicatrización de heridas. 1)
Cusumano et al. (2023) realizaron tratamiento 2RT en 3 pacientes con DHRD (41-46 años) e informaron un seguimiento de hasta 30 meses. 1) Los resultados principales fueron los siguientes:
Agudeza visual: Caso 1 mostró mejora de 2-10 letras
Sensibilidad del campo visual FDT: Caso 2 mostró mejora desde OD MD −12dB, Caso 3 desde OS MD −9dB
Amplitud del electrorretinograma de campo completo: Se confirmó aumento significativo en los Casos 1 y 2
Seguridad: No se observaron eventos adversos relacionados con el tratamiento (aunque apareció edema macular quístico a los 24 meses en el Caso 1)
Además, se observó una mejora del electrorretinograma específica de bastones (mejora del ERG de campo completo, sin cambios en el ERG multifocal), lo que sugiere un mecanismo de acción dirigido principalmente al sistema de bastones. 1) Además, se observó mejora funcional en el ojo contralateral no tratado en algunos casos, lo que sugiere la posibilidad de un efecto de inducción indirecta sistémica. 1)
Este informe se limita al nivel de evidencia de una serie de casos (3 casos); no se han realizado ensayos controlados aleatorios a gran escala. Establecer la eficacia y el protocolo óptimo para sesiones repetidas de láser sigue siendo un desafío futuro. 1)
Cusumano A, Falsini B, D’Ambrosio M, et al. Long-term structural and functional assessment of Doyne honeycomb retinal dystrophy following nanosecond 2RT laser treatment: a case series. Case Rep Ophthalmol. 2023;14:626-639.
Parameswarappa DC, Rani PK. Utility of pattern recognition and multimodal imaging in the diagnosis and management of doyne honeycomb retinal dystrophy complicated with type one choroidal neovascular membrane. BMJ Case Rep. 2021;14:e237635.
Tsang SH, Sharma T. Doyne Honeycomb Retinal Dystrophy (Malattia Leventinese, Autosomal Dominant Drusen). Adv Exp Med Biol. 2018;1085:97-102. PMID: 30578491.
Copia el texto del artículo y pégalo en el asistente de IA que prefieras.
Artículo copiado al portapapeles
Abre un asistente de IA abajo y pega el texto copiado en el chat.