دیستروفی شبکیه لانه زنبوری دویین (Doyne Honeycomb Retinal Dystrophy; DHRD) یک دیستروفی شبکیه با وراثت اتوزومال غالب است که با رسوبات سفید رنگ شبه دروزن به صورت شعاعی در قطب خلفی و اطراف پاپی مشخص میشود. همچنین با نامهای «مالاتیا لونتینزه (Malattia Leventinese; MLVT)» و «دروزن غالب خانوادگی» شناخته میشود. شماره OMIM #126600 است. 1)
در سال ۱۸۹۹، رابرت دویین، چشمپزشک بریتانیایی، این یافته را در چهار خواهر مشاهده و برای اولین بار به عنوان «الگوی لانه زنبوری» توصیف کرد. در سال ۱۹۲۵، وگت سوئیسی همان فنوتیپ را در دره لونتین گزارش و آن را «مالاتیا لونتینزه» نامید. در سال ۱۹۹۹، استون و همکاران جهش یکسان (R345W) در ژن EFEMP1 را در هر دو خانواده DHRD و MLVT شناسایی کردند و ثابت کردند که این دو یک بیماری هستند.
ژن مسئول EFEMP1 (پروتئین ماتریکس خارج سلولی حاوی فایبولین-1 شبه EGF) واقع در کروموزوم 2p16.1 است که پروتئین fibulin 3 را کد میکند. 2) این جهش missense Arg345Trp (R345W) باعث رسوب غیرطبیعی بین غشای بروخ و اپیتلیوم رنگدانه شبکیه (RPE) شده و منجر به تشکیل دروزن میشود. 1) با دژنراسیون ماکولای وابسته به سن در دروزن، ضخیم شدن غشای بروخ، آتروفی RPE و فعال شدن کمپلمان پاتوفیزیولوژی مشترک دارد. 1)
Qتفاوت DHRD و دژنراسیون ماکولای وابسته به سن چیست؟
A
دژنراسیون ماکولای وابسته به سن یک بیماری چندعاملی با علت اصلی افزایش سن است که در افراد مسن بروز میکند. در مقابل، DHRD ناشی از یک جهش تکژنی (R345W در EFEMP1) است، علائم در سنین جوانی تا میانسالی (دهه ۴۰ تا ۵۰) ظاهر میشود، سابقه خانوادگی دارد و به صورت اتوزومال غالب منتقل میشود. توزیع دروزن به صورت شعاعی در قطب خلفی و اطراف پاپی مشخصه آن است. از آنجا که هر دو مسیر پاتوفیزیولوژیک مشترک شامل تشکیل دروزن، تغییرات غشای بروخ و آتروفی RPE دارند، DHRD به عنوان مدل تحقیقاتی برای دژنراسیون ماکولای وابسته به سن مورد توجه است.
دروزنهای ناحیه خلفی و اطراف دیسک بینایی: دروزنهای زودرس در امتداد قوس عروقی. آرایش شعاعی مشخصه است.1)
دو نوع دروزن: دو نوع دروزن دیده میشود: دروزنهای گرد بزرگ و دروزنهای شعاعی کوچک.2)
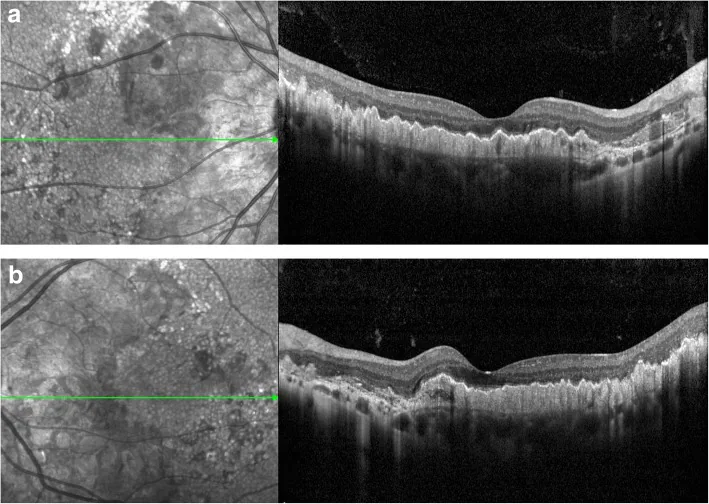
یافتههای OCT: تغییرات منتشر در کمپلکس RPE/غشای بروخ را نشان میدهد. لایه حسی عصبی نسبتاً حفظ میشود. PED شبهدروزن و علامت لایه دوتایی تأیید میشود.1)2)
یافتههای مرحله پیشرفته
همجوشی دروزن و تغییرات رنگدانهای: دروزنها به هم میپیوندند و ناهنجاری رنگدانهای در RPE ایجاد میشود.2)
آتروفی و اسکار RPE: ممکن است به آتروفی جغرافیایی مرکزی پیشرفت کند.2)
عارضه CNVM: نادر است اما علت اصلی کاهش بینایی است. ممکن است PED فیبروواسکولار ایجاد شود.2)
کاهش دامنه الکترورتینوگرام: در الکترورتینوگرام میدان کامل، کاهش دامنه تأیید میشود. 1)
ارزیابی با تصویربرداری چندوجهی برای تشخیص و پیگیری مفید است. 2) در خودفلورسانس فوندوس (FAF)، ناحیه دروزن دارای خودفلورسانس بالا و ناحیه آتروفی RPE دارای خودفلورسانس پایین است. 2) در آزمون آستانه فلیکر (FDT)، کاهش حساسیت میدان بینایی تشخیص داده میشود. 1)
Qآیا شدت علائم در افراد مختلف متفاوت است؟
A
حتی در یک خانواده، شدت بیماری متفاوت است. در برخی موارد، بینایی خوب تا مراحل پیشرفته حفظ میشود، در حالی که در موارد همراه با CNVM، کاهش شدید بینایی ممکن است رخ دهد. تشخیص زودهنگام CNVM از طریق معاینات منظم فوندوس اهمیت دارد.
DHRD یک بیماری ارثی اتوزومال غالب ناشی از جهش در یک ژن واحد است. اطلاعات ژنتیکی در زیر آورده شده است.
خلاصهای از اطلاعات ژنتیکی در زیر ارائه شده است.
مورد
محتوا
ژن عامل
EFEMP1 (2p16.1)
جهش
اگزون 10 · R345W
الگوی وراثت
اتوزومال غالب
پروتئین کدشونده
فیبولین 3
EFEMP1 پروتئین ماتریکس خارج سلولی شبه فیبریلین حاوی EGF 1 را کد میکند و به عنوان یک جزء ماتریکس خارج سلولی غشای بروخ عمل میکند. جهش R345W باعث تاخوردگی غیرطبیعی پروتئین شده و رسوبات لایه پایه بین RPE و غشای بروخ تجمع مییابند. 1) همچنین تصور میشود که جهش EFEMP1 با مهار سیگنال EGFR، CES1 درگیر در خروج کلسترول را مهار کرده و تجمع لیپید را تسریع میکند. در مدلهای موشی دژنراسیون ماکولای وابسته به سن و DHRD، افزایش فعال شدن کمپلمان نیز مشاهده میشود. 1)
تشخیص DHRD با تشخیص الگوی توزیع مشخص دروزن، شروع در سنین پایین و سابقه خانوادگی آغاز میشود. 2) ترکیبی از ارزیابی با تصویربرداری چندوجهی و آزمایش ژنتیکی برای تشخیص قطعی مفید است. 2)
تشخیص الگو: شروع در سنین جوانی (40-50 سالگی)، درگیری دوطرفه و متقارن، و توزیع شعاعی دروسن در قطب خلفی و اطراف دیسک بینایی سرنخهای تشخیصی هستند. 2)
OCT: تغییرات کمپلکس RPE/غشای بروخ، PED شبهدروسن، علامت لایه دوتایی، و حفظ لایه حسی عصبی را تأیید کنید. 2)
تصویربرداری خودفلورسانس فوندوس (FAF): خودفلورسانس بالای دروسن و الگوی خودفلورسانس پایین در نواحی آتروفی RPE را ارزیابی کنید. 2)
آزمایش ژنتیکی: توالییابی نسل بعدی با استفاده از NextSeq 550 (Illumina) برای تأیید جهش EFEMP1 (پوشش ≥20×). بر اساس راهنمای ACMG، c.1033C>T (R345W) به عنوان جهش بیماریزا (معیارهای PM2، PP3، PP5) طبقهبندی میشود. 1)
در صورت وجود علائم بالینی معمول (شروع در سنین پایین، دروزن قطبی خلفی دوطرفه متقارن، سابقه خانوادگی)، تشخیص بالینی بدون آزمایش ژنتیکی امکانپذیر است. با این حال، برای تشخیص قطعی، غربالگری خانواده و مشاوره ژنتیکی، تأیید جهش R345W در EFEMP1 توصیه میشود.
تزریق داخل زجاجیهای ضد VEGF یک گزینه درمانی برای موارد همراه با CNVM است. تزریق داخل زجاجیهای رانیبیزوماب (0.5 میلیگرم) استفاده میشود و پس از تزریق، بهبود بینایی و ناپدید شدن جداشدگی سروزی شبکیه (SRF) گزارش شده است. 2)
Parameswarappa و Rani یک زن 44 ساله (DHRD) را گزارش کردند که با غشای نئوواسکولار کوروئیدال نوع 1 همراه بود و پس از یک تزریق رانیبیزوماب، BCVA از 20/40 به 20/30 بهبود یافت و SRF ناپدید شد. 2) همچنین گزارشهایی از درمان CNVM با درمان فتودینامیک (PDT·verteporfin) وجود دارد. 2)
Qآیا میتوان درمان با لیزر نانوثانیه (2RT) را دریافت کرد؟
A
2RT (لیزر پالس نانوثانیه) به عنوان یک درمان جدید برای DHRD در گزارشهای موردی بهبود عملکرد را نشان داده است، اما در حال حاضر یک درمان تحقیقاتی است و به عنوان درمان استاندارد تثبیت نشده است. برای جزئیات به بخش «آخرین تحقیقات و چشماندازهای آینده» مراجعه کنید.
EFEMP1 (پروتئین ماتریکس خارج سلولی حاوی فیبولین-1 شبه EGF) به عنوان یک جزء ماتریکس خارج سلولی غشای بروخ عمل میکند.1) جهش R345W باعث تاخوردگی غیرطبیعی پروتئین شده و رسوبات لایه پایه بین RPE و غشای بروخ تجمع مییابد.1) این رسوبات زمینهساز تشکیل دروزن هستند.
ضخیم شدن غشای بروخ: کاهش نفوذپذیری به دلیل تغییرات ساختاری غشا
آتروفی RPE: دژنراسیون اپیتلیوم رنگدانهای به دلیل اختلال تغذیه
فعال شدن کمپلمان: افزایش فعالیت سیستم کمپلمان در مدلهای موشی هر دو بیماری تأیید شده است
به عنوان تأثیر بر سیگنالدهی EGFR، جهش EFEMP1 R345W مسیر EGFR را بیش از حد مهار کرده و بیان CES1 را که در دفع کلسترول نقش دارد، کاهش میدهد. این امر تجمع لیپیدها را تسریع کرده و منجر به تشکیل دروزن میشود.
در بررسیهای الکتروفیزیولوژیک، کاهش دامنه در الکترورتینوگرافی میدان کامل مشاهده میشود که نشاندهنده اختلال عملکرد هر دو نوع سلول استوانهای و مخروطی است.1) در آزمون میدان بینایی FDT، کاهش قابل توجهی در حساسیت میدان بینایی تأیید میشود.1)
Qچرا DHRD برای تحقیقات دژنراسیون ماکولای وابسته به سن مفید است؟
A
DHRD در حالی که مسیرهای مولکولی و پاتولوژیک یکسانی با دژنراسیون ماکولای وابسته به سن (تغییرات غشای بروخ، آتروفی RPE و فعال شدن کمپلمان) دارد، به دلیل یک جهش ژنی واحد (R345W) ایجاد میشود، بنابراین میتوان روابط علّی پاتولوژی را به وضوح تحلیل کرد. دژنراسیون ماکولای وابسته به سن یک بیماری چندعاملی ژنومی-محیطی است و تحلیل مکانیسم آن دشوار است، اما مدل DHRD سیستمی مناسب برای مطالعه مکانیسمهای مشترک آن فراهم میکند.
7. تحقیقات جدید و چشماندازهای آینده (گزارشهای مرحله تحقیقاتی)
2RT (درمان شبکیه در 2 دقیقه) یک روش درمانی غیرتهاجمی با استفاده از لیزر پالس نانوثانیه با انرژی بسیار کم (قطر 400 میکرومتر، 3 نانوثانیه، 532 نانومتر، 0.15 تا 0.45 میلیژول) است. مکانیسم فرضی آن شامل دبریدمان اپیتلیوم رنگدانهدار شبکیه (RPE) و ترویج بازسازی لایهها از طریق پاسخ ترمیم زخم است. 1)
کوزومانو و همکاران (2023) درمان 2RT را بر روی 3 بیمار مبتلا به DHRD (سن 41 تا 46 سال) انجام دادند و پیگیری تا 30 ماه را گزارش کردند. 1) نتایج اصلی به شرح زیر بود:
حدت بینایی: در مورد 1 بهبود 2 تا 10 حرف مشاهده شد
حساسیت میدان بینایی FDT: در مورد 2 بهبود از MD −12 دسیبل در چشم راست و در مورد 3 بهبود از MD −9 دسیبل در چشم چپ مشاهده شد
دامنه الکترورتینوگرافی تمام میدان: در موارد 1 و 2 افزایش معنیدار تأیید شد
ایمنی: هیچ عارضه جانبی مرتبط با درمان مشاهده نشد (اگرچه در مورد 1 در ماه 24 ادم ماکولای کیستیک ظاهر شد)
همچنین بهبود الکترورتینوگرافی اختصاصی سلولهای استوانهای (بهبود ERG تمام میدان بدون تغییر در الکترورتینوگرافی چندکانونی) مشاهده شد که مکانیسم اثر عمدتاً بر سیستم استوانهای را نشان میدهد. 1) علاوه بر این، در برخی موارد بهبود عملکرد در چشم مقابل درماننشده نیز دیده شد که احتمال اثر القایی غیرمستقیم سیستمیک را مطرح میکند. 1)
این گزارش تنها در سطح شواهد یک سری موارد (3 بیمار) است و کارآزمایی بالینی تصادفیسازی شده در مقیاس بزرگ انجام نشده است. اثربخشی جلسات مکرر لیزر و تعیین پروتکل بهینه از چالشهای آینده محسوب میشود. 1)
Cusumano A, Falsini B, D’Ambrosio M, et al. Long-term structural and functional assessment of Doyne honeycomb retinal dystrophy following nanosecond 2RT laser treatment: a case series. Case Rep Ophthalmol. 2023;14:626-639.
Parameswarappa DC, Rani PK. Utility of pattern recognition and multimodal imaging in the diagnosis and management of doyne honeycomb retinal dystrophy complicated with type one choroidal neovascular membrane. BMJ Case Rep. 2021;14:e237635.
Tsang SH, Sharma T. Doyne Honeycomb Retinal Dystrophy (Malattia Leventinese, Autosomal Dominant Drusen). Adv Exp Med Biol. 2018;1085:97-102. PMID: 30578491.
متن مقاله را کپی کنید و در دستیار هوش مصنوعی دلخواه خود بچسبانید.
مقاله در کلیپبورد کپی شد
یکی از دستیارهای هوش مصنوعی زیر را باز کنید و متن کپیشده را در کادر گفتگو بچسبانید.