La dystrophie rétinienne en rayon de miel de Doyne (Doyne Honeycomb Retinal Dystrophy ; DHRD) est une dystrophie rétinienne autosomique dominante caractérisée par des dépôts blancs ressemblant à des drusen disposés radialement autour de la région postérieure et de la papille. Elle est également appelée « Malattia Leventinese (MLVT) » ou « drusen familiales dominantes ». Le numéro OMIM est #126600. 1)
En 1899, l’ophtalmologiste britannique Robert Doyne a observé cette manifestation chez quatre sœurs et l’a décrite pour la première fois comme un « motif en nid d’abeille ». En 1925, le Suisse Vogt a rapporté le même phénotype dans la vallée de Leventine et l’a nommé « Malattia Leventinese ». En 1999, Stone et al. ont identifié la même mutation (R345W) du gène EFEMP1 dans les deux familles DHRD et MLVT, prouvant qu’il s’agit de la même maladie.
Le gène responsable est EFEMP1 (EGF-containing fibulin-like extracellular matrix protein 1), situé sur le chromosome 2p16.1, qui code la protéine fibuline 3. 2) Cette mutation faux-sens Arg345Trp (R345W) provoque des dépôts anormaux entre la membrane de Bruch et l’épithélium pigmentaire rétinien (EPR), entraînant la formation de drusen. 1) La dégénérescence maculaire liée à l’âge et la dystrophie réticulée de Doyne partagent des mécanismes pathologiques communs : drusen, épaississement de la membrane de Bruch, atrophie de l’EPR et activation du complément. 1)
QQuelle est la différence entre la DHRD et la dégénérescence maculaire liée à l'âge ?
A
La dégénérescence maculaire liée à l’âge est une maladie multifactorielle principalement due au vieillissement, survenant chez les personnes âgées. En revanche, la DHRD est causée par une mutation génétique unique, la mutation R345W du gène EFEMP1, avec des symptômes apparaissant chez les jeunes et les adultes d’âge moyen (40-50 ans), suivant un mode de transmission autosomique dominant avec des antécédents familiaux. La distribution des drusen est caractéristique, avec un arrangement radial autour du pôle postérieur et de la papille optique. Les deux pathologies partagent des voies pathogéniques communes : formation de drusen, modifications de la membrane de Bruch et atrophie de l’EPR, ce qui fait de la DHRD un modèle d’étude pour la dégénérescence maculaire liée à l’âge.
Scotome : apparaît comme un scotome central ou paracentral.
Dyschromatopsie : observée au stade avancé.
Baisse de l’acuité visuelle : une baisse marquée survient en cas de complication par une CNVM. Au stade avancé, l’acuité visuelle peut devenir inférieure à 20/200. 2)
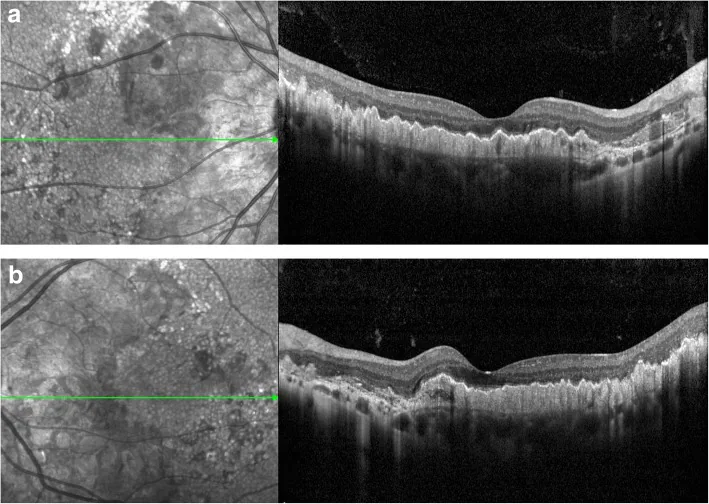
Drusen du pôle postérieur et péripapillaires : drusen précoces le long de l’arcade vasculaire. Disposition radiaire caractéristique. 1)
Deux types de drusen : on observe deux types : de grands drusen ronds et de petits drusen radiaires. 2)
Signes OCT : modifications diffuses du complexe RPE/membrane de Bruch. La couche neurosensorielle est relativement préservée. On observe un PED de type drusen et un signe de double couche. 1)2)
Signes de stade avancé
Fusion des drusen et altérations pigmentaires : les drusen fusionnent et des anomalies pigmentaires de l’EPR apparaissent. 2)
Atrophie et cicatrisation de l’EPR : peut évoluer vers une atrophie géographique centrale. 2)
Complication par CNVM : rare mais principale cause de baisse de l’acuité visuelle. Peut se présenter sous forme de décollement pigmentaire fibrovasculaire. 2)
Réduction de l’amplitude de l’électrorétinogramme : Une réduction de l’amplitude est confirmée à l’électrorétinogramme champ total. 1)
L’évaluation par imagerie multimodale est utile pour le diagnostic et le suivi. 2) En autofluorescence du fond d’œil (FAF), on observe une hyper-autofluorescence au niveau des drusen et une hypo-autofluorescence dans les zones d’atrophie de l’EPR. 2) Le test de seuil de flicker (FDT) détecte une diminution de la sensibilité du champ visuel. 1)
QExiste-t-il des différences individuelles dans la sévérité des symptômes ?
A
Même au sein d’une même famille, la sévérité varie. Certains patients conservent une bonne acuité visuelle jusqu’à un stade avancé, tandis que d’autres, en cas de complication par une CNVM, peuvent présenter une baisse rapide de l’acuité visuelle. Un dépistage précoce de la CNVM par des examens réguliers du fond d’œil est important.
La DHRD est une maladie autosomique dominante due à une mutation génétique unique. Les informations génétiques sont présentées ci-dessous.
Un résumé des informations génétiques est présenté ci-dessous.
Élément
Contenu
Gène causal
EFEMP1 (2p16.1)
Mutation
Exon 10, R345W
Mode de transmission
Autosomique dominant
Protéine codée
Fibuline 3
EFEMP1 code la protéine 1 de la matrice extracellulaire fibrineuse contenant EGF et agit comme un composant de la matrice extracellulaire de la membrane de Bruch. La mutation R345W provoque un mauvais repliement de la protéine, entraînant l’accumulation de dépôts de couche basale entre l’EPR et la membrane de Bruch. 1) De plus, la mutation EFEMP1 inhiberait la CES1 impliquée dans l’efflux de cholestérol via la suppression du signal EGFR, favorisant ainsi l’accumulation de lipides. Dans les modèles murins de dégénérescence maculaire liée à l’âge et de DHRD, une activation accrue du complément est également observée. 1)
Le diagnostic de la DHRD commence par la reconnaissance de l’apparition juvénile, des antécédents familiaux et de la distribution caractéristique des drusen. 2) La combinaison d’une évaluation par imagerie multimodale et de tests génétiques est utile pour un diagnostic définitif. 2)
Reconnaissance de motifs : l’apparition chez le sujet jeune (40-50 ans), la bilatéralité symétrique et la distribution radiaire des drusen autour du pôle postérieur et de la papille sont des indices diagnostiques. 2)
OCT : confirmer les altérations du complexe RPE/membrane de Bruch, les PED de type drusen, le signe de la double couche et la préservation de la couche neurosensorielle. 2)
Autofluorescence du fond d’œil (FAF) : évaluer l’hyperautofluorescence des drusen et l’hypoautofluorescence des zones d’atrophie de l’EPR. 2)
Test génétique : séquençage de nouvelle génération avec NextSeq 550 (Illumina) pour confirmer la mutation EFEMP1 (couverture ≥20×). Selon les directives de l’ACMG, c.1033C>T (R345W) est classée comme mutation pathogène (critères PM2, PP3, PP5). 1)
Il est important de différencier les maladies présentant des signes du fond d’œil similaires.
Maladie
Caractéristiques
Points de différenciation
Dégénérescence maculaire liée à l’âge
Apparition tardive, multifactorielle
Âge d’apparition, pas d’antécédents familiaux
Dystrophie de Sorsby
Mutation TIMP3
Drusen réticulaires
Maladie de Stargardt
Mutation ABCA4
Taches maculaires
MPGN de type II
Maladie systémique
Avec anomalie de la fonction rénale
QUn test génétique est-il toujours nécessaire pour un diagnostic définitif ?
A
Lorsque les signes cliniques sont typiques (apparition précoce, drusen symétriques du pôle postérieur dans les deux yeux, antécédents familiaux), un diagnostic clinique est possible sans test génétique. Cependant, pour un diagnostic définitif, un dépistage familial et un conseil génétique, la confirmation de la mutation R345W du gène EFEMP1 est recommandée.
Il n’existe actuellement aucun traitement modificateur de la maladie établi pour la DHRD. La stratégie thérapeutique diffère selon la présence ou non d’une CNVM associée.
Surveillance régulière : examen du fond d’œil et OCT pour détecter précocement une CNVM.
Réadaptation visuelle : en cas de baisse significative de l’acuité visuelle au stade avancé, une réadaptation visuelle et un entraînement à la mobilité sont proposés. 2)
L’injection intravitréenne d’anti-VEGF est une option thérapeutique pour les cas compliqués de CNVM. L’injection intravitréenne de ranibizumab (0,5 mg) est utilisée, et une amélioration de l’acuité visuelle ainsi qu’une disparition du décollement séreux de la rétine (SRF) ont été rapportées après l’injection. 2)
Parameswarappa et Rani ont rapporté le cas d’une femme de 44 ans (DHRD) présentant une membrane néovasculaire choroïdienne de type 1. Après une seule injection de ranibizumab, la meilleure acuité visuelle corrigée (BCVA) est passée de 20/40 à 20/30, et le SRF a disparu. 2) La thérapie photodynamique (PDT, vertéporfine) a également été rapportée pour le traitement de la CNVM. 2)
QPuis-je recevoir un traitement au laser nanoseconde (2RT) ?
A
Le 2RT (laser à impulsions nanosecondes) a été rapporté dans des cas cliniques comme améliorant la fonction visuelle pour le DHRD, mais il s’agit actuellement d’un traitement expérimental non établi comme standard. Veuillez vous référer à la section « Recherches récentes et perspectives futures » pour plus de détails.
EFEMP1 (protéine 1 de la matrice extracellulaire de type fibuline contenant un domaine EGF) fonctionne comme un composant de la matrice extracellulaire de la membrane de Bruch. 1) La mutation R345W entraîne un mauvais repliement de la protéine, ce qui provoque l’accumulation de dépôts de couche basale entre l’EPR et la membrane de Bruch. 1) Ces dépôts constituent la base de la formation de drusen.
La DHRD et la dégénérescence maculaire liée à l’âge partagent les voies pathologiques suivantes. 1)
Formation de drusen : dépôt de substances anormales dans la membrane de Bruch
Épaississement de la membrane de Bruch : diminution de la perméabilité due à des modifications structurelles de la membrane
Atrophie de l’EPR : dégénérescence de l’épithélium pigmentaire due à des troubles nutritionnels
Activation du complément : une hyperactivation du système du complément a été confirmée dans des modèles murins des deux maladies.
Concernant l’impact sur la signalisation EGFR, la mutation EFEMP1 R345W supprime excessivement la voie EGFR et réduit l’expression de CES1, impliquée dans l’efflux de cholestérol. Cela favorise l’accumulation lipidique et conduirait à la formation de drusen.
Les études électrophysiologiques montrent une réduction d’amplitude à l’électrorétinogramme champ total, suggérant un dysfonctionnement des bâtonnets et des cônes. 1) Les tests de champ visuel FDT confirment une diminution significative de la sensibilité du champ visuel. 1)
QPourquoi la DHRD est-elle utile pour la recherche sur la dégénérescence maculaire liée à l'âge ?
A
La DHRD partage les mêmes voies moléculaires et pathologiques que la dégénérescence maculaire liée à l’âge (altérations de la membrane de Bruch, atrophie de l’EPR, activation du complément) mais est causée par une mutation génétique unique (R345W), ce qui permet d’analyser clairement les relations de cause à effet. Alors que la dégénérescence maculaire liée à l’âge est une maladie multifactorielle impliquant des facteurs génétiques et environnementaux, rendant l’analyse des mécanismes difficile, le modèle DHRD constitue un système adapté pour étudier ces mécanismes communs.
7. Recherches récentes et perspectives futures (rapports en phase de recherche)
La 2RT (traitement rétinien de 2 minutes) est une méthode non invasive utilisant un laser à impulsions nanosecondes de très faible énergie (diamètre 400 μm, 3 nanosecondes, 532 nm, 0,15-0,45 mJ). On suppose qu’elle favorise la régénération des couches via le débridement de l’EPR et la réponse de cicatrisation. 1)
Cusumano et al. (2023) ont traité 3 patients atteints de DHRD (âgés de 41 à 46 ans) par 2RT et ont rapporté un suivi allant jusqu’à 30 mois. 1) Les principaux résultats étaient les suivants :
Acuité visuelle : Amélioration de 2 à 10 lettres dans le cas 1
Sensibilité du champ visuel FDT : Amélioration à partir de OD MD −12 dB dans le cas 2 et de OS MD −9 dB dans le cas 3
Amplitude de l’électrorétinogramme plein champ : Augmentation significative confirmée dans les cas 1 et 2
Sécurité : Aucun événement indésirable lié au traitement n’a été observé (bien qu’un œdème maculaire cystoïde soit apparu à 24 mois dans le cas 1)
De plus, une amélioration de l’électrorétinogramme spécifique aux bâtonnets (amélioration de l’ERG plein champ, pas de changement à l’électrorétinogramme multifocal) a été observée, suggérant un mécanisme d’action ciblant principalement le système des bâtonnets. 1) En outre, des cas d’amélioration fonctionnelle ont été observés dans l’œil controlatéral non traité, ce qui suggère un possible effet d’induction indirecte systémique. 1)
Ce rapport se limite au niveau de preuve d’une série de cas (3 cas) et aucun essai contrôlé randomisé à grande échelle n’a été réalisé. L’établissement de l’efficacité et du protocole optimal pour les sessions laser répétées est considéré comme un défi futur. 1)
Cusumano A, Falsini B, D’Ambrosio M, et al. Long-term structural and functional assessment of Doyne honeycomb retinal dystrophy following nanosecond 2RT laser treatment: a case series. Case Rep Ophthalmol. 2023;14:626-639.
Parameswarappa DC, Rani PK. Utility of pattern recognition and multimodal imaging in the diagnosis and management of doyne honeycomb retinal dystrophy complicated with type one choroidal neovascular membrane. BMJ Case Rep. 2021;14:e237635.
Tsang SH, Sharma T. Doyne Honeycomb Retinal Dystrophy (Malattia Leventinese, Autosomal Dominant Drusen). Adv Exp Med Biol. 2018;1085:97-102. PMID: 30578491.
Copiez le texte de l'article et collez-le dans l'assistant IA de votre choix.
Article copié dans le presse-papiers
Ouvrez un assistant IA ci-dessous et collez le texte copié dans la conversation.