Die Doyne-Honigwaben-Netzhautdystrophie (Doyne Honeycomb Retinal Dystrophy; DHRD) ist eine autosomal-dominant vererbte Netzhautdystrophie, bei der weiße drusenartige Ablagerungen strahlenförmig um die hintere Polregion und die Papille angeordnet sind. Sie wird auch als Malattia Leventinese (MLVT) oder familiäre dominante Drusen bezeichnet. Die OMIM-Nummer ist #126600. 1)
Im Jahr 1899 beschrieb der britische Augenarzt Robert Doyne diesen Befund erstmals bei vier Schwestern und prägte den Begriff „Honigwabenmuster“. 1925 berichtete der Schweizer Vogt über denselben Phänotyp im Leventinatal und nannte ihn „Malattia Leventinese“. 1999 identifizierten Stone et al. in beiden Familien (DHRD und MLVT) dieselbe Mutation (R345W) im EFEMP1-Gen und bewiesen, dass es sich um dieselbe Erkrankung handelt.
Das verantwortliche Gen ist EFEMP1 (EGF-containing fibulin-like extracellular matrix protein 1) auf Chromosom 2p16.1, das für das Fibulin-3-Protein kodiert. 2) Diese Missense-Mutation Arg345Trp (R345W) führt zu abnormalen Ablagerungen zwischen der Bruch-Membran und dem retinalen Pigmentepithel (RPE), was zur Bildung von Drusen führt. 1) Die altersbedingte Makuladegeneration und Drusen, Bruch-Membran-Verdickung, RPE-Atrophie und Komplementaktivierung teilen gemeinsame pathologische Merkmale. 1)
QWie unterscheidet sich DHRD von der altersbedingten Makuladegeneration?
A
Die altersbedingte Makuladegeneration ist eine multifaktorielle Erkrankung, die hauptsächlich durch das Alter verursacht wird und bei älteren Menschen auftritt. Die DHRD hingegen wird durch eine einzelne Genmutation, die R345W-Mutation des EFEMP1-Gens, verursacht. Die Symptome treten im jungen bis mittleren Alter (40–50 Jahre) auf und folgen einem autosomal-dominanten Erbgang mit Familienanamnese. Die Drusenverteilung ist charakteristisch radial um den hinteren Pol und die Papille angeordnet. Da beide Erkrankungen gemeinsame pathologische Wege wie Drusenbildung, Veränderungen der Bruch-Membran und RPE-Atrophie teilen, gilt die DHRD auch als Forschungsmodell für die altersbedingte Makuladegeneration.
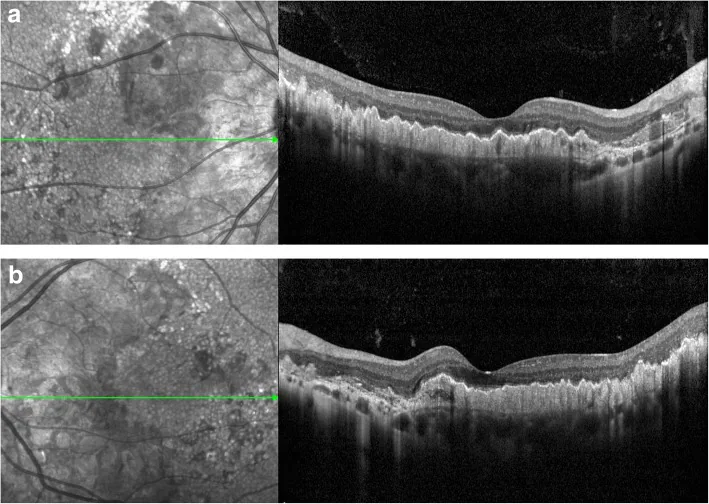
Kaiyan Zhang; Xuyang Sun; Yingying Chen; Qionglei Zhong; Lin Lin; Yuan Gao; Fanlin Hong. Doyne honeycomb retinal dystrophy/malattia leventinese induced by EFEMP1 mutation in a Chinese family. BMC Ophthalmol. 2018 Dec 12; 18:318. Figure 3. PMCID: PMC6292057. License: CC BY.
OCT-Scan. Eine hyperreflektive Verdickung unterhalb des RPE wurde festgestellt, begleitet von wellenförmiger Anhebung (a: rechtes Auge; b: linkes Auge).
Im Frühstadium verläuft die Erkrankung oft asymptomatisch. Ab dem 40. bis 50. Lebensjahr treten folgende Symptome auf.
Verschwommenes Sehen / unscharfes zentrales Sehen: Kann mit einer verminderten Kontrastwahrnehmung einhergehen. 1)
Metamorphopsie: Wird als Verzerrung im zentralen Gesichtsfeld wahrgenommen.
Skotom: Tritt als zentrales oder parazentrales Skotom auf.
Farbsehstörung: Tritt im fortgeschrittenen Stadium auf.
Sehverschlechterung: Bei Komplikation durch CNVM kommt es zu einer deutlichen Sehverschlechterung. Im fortgeschrittenen Stadium kann die Sehschärfe auf 20/200 oder weniger sinken. 2)
Drusen im hinteren Pol und peripapillär: Früh auftretende Drusen entlang des Gefäßbogens. Charakteristisch ist eine radiäre Anordnung. 1)
Zwei Drusenformen: Es werden zwei Typen unterschieden: große runde Drusen und kleine radiäre Drusen. 2)
OCT-Befunde: Diffuse Veränderungen des RPE-Bruch-Membran-Komplexes. Die neurosensorische Schicht bleibt relativ erhalten. Drusenartige PED und Double-Layer-Sign sind nachweisbar. 1)2)
Befunde im fortgeschrittenen Stadium
Drusenkonfluenz und Pigmentveränderungen: Drusen konfluieren, und es treten Pigmentanomalien im RPE auf. 2)
RPE-Atrophie und Narbenbildung: Kann zu einer geografischen Atrophie im Zentrum fortschreiten. 2)
CNVM-Komplikation: Selten, aber Hauptursache für Sehverschlechterung. Kann eine fibrovaskuläre PED aufweisen. 2)
Verminderte Amplitude im Elektroretinogramm: Im Ganzfeld-Elektroretinogramm wird eine verminderte Amplitude festgestellt. 1)
Die Bewertung mittels multimodaler Bildgebung ist für die Diagnose und Verlaufskontrolle nützlich. 2) In der Fundus-Autofluoreszenz (FAF) zeigt sich eine hohe Autofluoreszenz im Bereich der Drusen und eine niedrige Autofluoreszenz im Bereich der RPE-Atrophie. 2) Der Flimmerschwellentest (FDT) erfasst eine verminderte Gesichtsfeldempfindlichkeit. 1)
QGibt es individuelle Unterschiede im Schweregrad der Symptome?
A
Auch innerhalb derselben Familie gibt es Unterschiede im Schweregrad. Bei einigen Fällen bleibt bis zum fortgeschrittenen Stadium eine gute Sehschärfe erhalten, während es bei Fällen mit CNVM zu einem plötzlichen Sehverlust kommen kann. Regelmäßige augenärztliche Untersuchungen zur Früherkennung einer CNVM sind wichtig.
DHRD ist eine autosomal-dominante Erbkrankheit, die durch eine einzelne Genmutation verursacht wird. Nachfolgend sind die genetischen Informationen aufgeführt.
Eine Übersicht der genetischen Informationen ist unten dargestellt.
Parameter
Inhalt
Krankheitsgen
EFEMP1 (2p16.1)
Mutation
Exon 10 · R345W
Vererbungsmuster
autosomal-dominant
kodiertes Protein
Fibulin-3
EFEMP1 kodiert für EGF-enthaltendes Fibrillin-ähnliches extrazelluläres Matrixprotein 1 und fungiert als Bestandteil der extrazellulären Matrix der Bruch-Membran. Die R345W-Mutation führt zu einer Fehlfaltung des Proteins, wodurch sich Basallaminaablagerungen zwischen dem RPE und der Bruch-Membran ansammeln. 1) Zudem wird angenommen, dass die EFEMP1-Mutation durch Unterdrückung des EGFR-Signals die Cholesterinausscheidung hemmende CES1 blockiert und so die Lipidakkumulation fördert. In Mausmodellen der altersbedingten Makuladegeneration und der DHRD wird auch eine erhöhte Komplementaktivierung beobachtet. 1)
Die Diagnose der DHRD beginnt mit der Erkennung des frühen Erkrankungsalters, der Familienanamnese und des charakteristischen Verteilungsmusters der Drusen. 2) Die Kombination aus multimodaler Bildgebung und Gentests ist für die definitive Diagnose hilfreich. 2)
Mustererkennung: Frühes Erkrankungsalter (40–50 Jahre), bilaterale Symmetrie, radiale Drusenverteilung im hinteren Pol und um die Papille sind diagnostische Hinweise. 2)
OCT: Veränderungen des RPE/Bruch-Membran-Komplexes, drusenartige PED, Double-Layer-Sign und Erhalt der neurosensorischen Schicht bestätigen. 2)
Fundusautofluoreszenz (FAF): Bewertung des hyperautofluoreszenten Drusenmusters und des hypoautofluoreszenten Bereichs der RPE-Atrophie. 2)
Gentest: Next-Generation-Sequencing mit NextSeq 550 (Illumina) bestätigt die EFEMP1-Mutation (Coverage ≥20×). Gemäß ACMG-Richtlinien wird c.1033C>T (R345W) als pathogene Mutation (Kriterien PM2, PP3, PP5) eingestuft. 1)
Kein frühes Erkrankungsalter, keine Familienanamnese
Sorsby-Dystrophie
TIMP3-Mutation
Netzartige Drusen
Stargardt-Krankheit
ABCA4-Mutation
Makuläre Flecken
MPGN Typ II
Systemische Erkrankung
Mit Nierenfunktionsstörung
QIst ein Gentest für die definitive Diagnose zwingend erforderlich?
A
Bei typischen klinischen Befunden (frühes Erkrankungsalter, bilaterale symmetrische Drusen der hinteren Polregion, Familienanamnese) ist eine klinische Diagnose auch ohne Gentest möglich. Für eine definitive Diagnose, Familienuntersuchung und genetische Beratung wird jedoch der Nachweis der EFEMP1-R345W-Mutation empfohlen.
Intravitreale Anti-VEGF-Injektion ist eine Behandlungsoption bei CNVM-Komplikationen. Intravitreale Injektion von Ranibizumab (0,5 mg) wird eingesetzt, und nach der Injektion wurden eine Verbesserung der Sehschärfe und das Verschwinden der serösen Netzhautablösung (SRF) berichtet. 2)
Parameswarappa und Rani berichteten über eine 44-jährige Frau (DHRD) mit einer choroidalen Neovaskularisationsmembran vom Typ 1, bei der nach einer einzigen Ranibizumab-Injektion der bestkorrigierte Visus (BCVA) von 20/40 auf 20/30 anstieg und die SRF verschwand. 2) Es gibt auch Berichte über die Behandlung von CNVM mit photodynamischer Therapie (PDT, Verteporfin). 2)
QKann eine Nanosekunden-Laserbehandlung (2RT) durchgeführt werden?
A
Die 2RT (Nanosekunden-Pulslaser) wird in Fallberichten als neue Behandlung für DHRD mit funktioneller Verbesserung beschrieben, ist jedoch derzeit noch eine experimentelle Therapie und nicht als Standardbehandlung etabliert. Einzelheiten finden Sie im Abschnitt „Neueste Forschung und zukünftige Perspektiven“.
6. Pathophysiologie und detaillierter Krankheitsmechanismus
EFEMP1 (EGF-containing fibulin-like extracellular matrix protein 1) fungiert als Bestandteil der extrazellulären Matrix der Bruch-Membran. 1) Die R345W-Mutation führt zu einer abnormalen Proteinfaltung, was zur Ansammlung von Basallaminaablagerungen zwischen RPE und Bruch-Membran führt. 1) Diese Ablagerungen bilden die Grundlage für die Drusenbildung.
Drusenbildung: Ablagerung abnormaler Substanzen in der Bruch-Membran
Verdickung der Bruch-Membran: Verminderte Durchlässigkeit durch strukturelle Veränderungen der Membran
RPE-Atrophie: Degeneration des Pigmentepithels aufgrund von Ernährungsstörungen
Komplementaktivierung: In Mausmodellen beider Erkrankungen wurde eine erhöhte Komplementaktivität nachgewiesen.
Hinsichtlich des EGFR-Signalwegs führt die EFEMP1 R345W-Mutation zu einer übermäßigen Hemmung des EGFR-Signalwegs und verringert die Expression von CES1, das am Cholesterinausstoß beteiligt ist. Dies fördert die Lipidakkumulation und führt vermutlich zur Drusenbildung.
Elektrophysiologische Untersuchungen zeigen eine verminderte Amplitude im Ganzfeld-Elektroretinogramm, was auf eine Funktionsstörung sowohl der Stäbchen als auch der Zapfen hindeutet. 1)FDT-Gesichtsfelduntersuchungen zeigen eine signifikante Abnahme der Gesichtsfeldempfindlichkeit. 1)
QWarum ist DHRD für die Altersabhängige Makuladegenerationsforschung nützlich?
A
DHRD weist dieselben molekularen und pathologischen Signalwege (Bruch-Membran-Veränderungen, RPE-Atrophie, Komplementaktivierung) wie die altersabhängige Makuladegeneration auf, wird jedoch durch eine einzelne Genmutation (R345W) verursacht, sodass die kausalen Zusammenhänge der Pathogenese klar analysiert werden können. Die altersabhängige Makuladegeneration ist eine multifaktorielle Erkrankung mit genomischen und umweltbedingten Faktoren, deren Mechanismus schwer zu analysieren ist, aber das DHRD-Modell bietet ein geeignetes System zur Untersuchung dieser gemeinsamen Mechanismen.
7. Aktuelle Forschung und zukünftige Perspektiven (Forschungsstadium)
Die 2RT (2-Minuten-Retina-Behandlung) ist eine nicht-invasive Behandlungsmethode, die einen ultraniedrigen Energie-Nanosekunden-Pulslaser (400 μm Durchmesser, 3 Nanosekunden, 532 nm, 0,15–0,45 mJ) verwendet. Es wird angenommen, dass der Mechanismus die RPE-Debridement und die Förderung der Schichtneubildung durch Wundheilungsreaktionen umfasst. 1)
Cusumano et al. (2023) führten eine 2RT-Behandlung bei 3 DHRD-Patienten (41–46 Jahre) durch und berichteten über ein Follow-up von bis zu 30 Monaten. 1) Die Hauptergebnisse waren wie folgt:
Sehschärfe: Bei Fall 1 wurde eine Verbesserung um 2–10 Buchstaben festgestellt
FDT-Gesichtsfeldempfindlichkeit: Bei Fall 2 eine Verbesserung von OD MD −12 dB, bei Fall 3 eine Verbesserung von OS MD −9 dB
Ganzfeld-Elektroretinogramm-Amplitude: Bei Fall 1 und 2 wurde ein signifikanter Anstieg bestätigt
Sicherheit: Es wurden keine behandlungsbedingten unerwünschten Ereignisse festgestellt (allerdings trat bei Fall 1 nach 24 Monaten ein zystoides Makulaödem auf)
Zudem wurde eine stäbchenspezifische Verbesserung des Elektroretinogramms (Verbesserung im Ganzfeld-ERG, keine Veränderung im multifokalen ERG) festgestellt, was auf einen Wirkmechanismus hindeutet, der hauptsächlich auf das Stäbchensystem abzielt. 1) Darüber hinaus gab es Fälle, bei denen auch das unbehandelte Partnerauge eine funktionelle Verbesserung zeigte, was auf eine mögliche systemische indirekte Induktionswirkung hindeutet. 1)
Cusumano A, Falsini B, D’Ambrosio M, et al. Long-term structural and functional assessment of Doyne honeycomb retinal dystrophy following nanosecond 2RT laser treatment: a case series. Case Rep Ophthalmol. 2023;14:626-639.
Parameswarappa DC, Rani PK. Utility of pattern recognition and multimodal imaging in the diagnosis and management of doyne honeycomb retinal dystrophy complicated with type one choroidal neovascular membrane. BMJ Case Rep. 2021;14:e237635.
Tsang SH, Sharma T. Doyne Honeycomb Retinal Dystrophy (Malattia Leventinese, Autosomal Dominant Drusen). Adv Exp Med Biol. 2018;1085:97-102. PMID: 30578491.
Kopieren Sie den Artikeltext und fügen Sie ihn in den KI-Assistenten Ihrer Wahl ein.
Artikel in die Zwischenablage kopiert
Öffnen Sie unten einen KI-Assistenten und fügen Sie den kopierten Text in den Chat ein.