La sindrome di Brown-McLean è una malattia in cui si verifica edema nella cornea periferica inferiore (a 2-3 mm dal limbo) che progredisce circonferenzialmente. La cornea centrale è preservata nella maggior parte dei casi.
Nel 1969, Brown e McLean riportarono per la prima volta un «edema corneale periferico dopo estrazione di cataratta». Successivamente, la malattia fu denominata e rimane tale fino ad oggi.
È una malattia relativamente rara, classicamente riportata più spesso in occhi afachici dopo estrazione intracapsulare della cataratta (ICCE). È noto che si sviluppa in media 6-16 anni dopo l’intervento. La maggior parte dei pazienti sono anziani, ma sono stati riportati casi anche in giovani di 12 anni.
Esistono casi di miopia elevata associati alla sindrome di Brown-McLean, con una prevalenza riportata del 40-61%.
QQuanto tempo dopo l'intervento di cataratta si sviluppa?
A
Si sviluppa dopo un lungo periodo di latenza di 6-16 anni in media dopo l’intervento. Pertanto, durante la visita ambulatoriale di pazienti anziani con storia di intervento di cataratta, è importante controllare lo stato della cornea periferica anche molto tempo dopo l’operazione.
Guedes J, et al. Biomechanical and tomographic findings in Brown-McLean syndrome. Am J Ophthalmol Case Rep. 2024. Figure 6. PMCID: PMC11359765. License: CC BY.
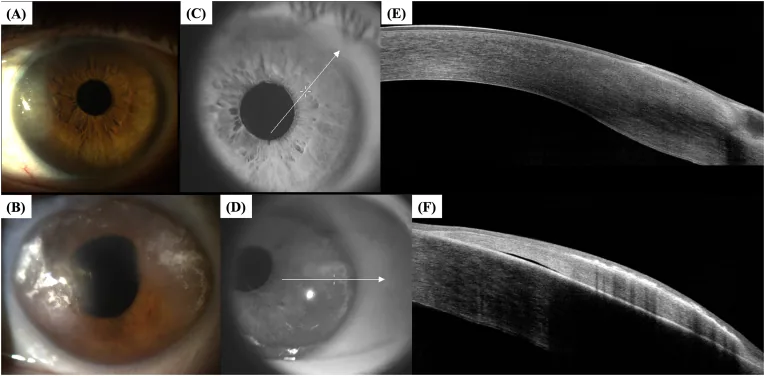
Fotografie al microscopio a lampada a fessura del segmento anteriore dell’occhio destro (A) e dell’occhio sinistro (B) del caso 3, e immagini di tomografia a coerenza ottica del segmento anteriore (AS-OCT) dell’occhio destro (E) e dell’occhio sinistro (F) acquisite in base alle linee di scansione (C) e (D). Corrispondono all’edema corneale periferico trattato nella sezione «2. Principali sintomi e segni clinici».
Di solito asintomatico. Quando compaiono sintomi, i seguenti sono comuni:
Sensazione di corpo estraneo: può essere avvertita con la progressione dell’edema periferico.
Fotofobia (fastidio alla luce) : osservata quando l’infiammazione o l’edema peggiorano.
Riduzione dell’acuità visiva : finché la cornea centrale è preservata, l’impatto sulla vista è minimo. Nei casi gravi con scompenso della cornea centrale, la riduzione della vista diventa marcata.
L’esame con lampada a fessura è centrale per la diagnosi. I reperti caratteristici sono mostrati di seguito.
Reperti periferici
Edema corneale periferico : inizia inizialmente nella parte periferica inferiore, compreso il limbo, e progredisce in modo circonferenziale. La cornea superiore è generalmente preservata, ma esistono rari casi in cui l’edema inizia dall’alto.
Pigmentazione endoteliale bruno-arancio: pigmentazione caratteristica dell’endotelio corneale nell’area dell’edema. Si ritiene causata da un trauma endoteliale intermittente dovuto a iridodonesi.
Atrofia dell’iride: è stata riportata un’atrofia dell’ire direttamente al di sotto dell’area di edema corneale.
Reperti centrali e generali
Mantenimento della trasparenza corneale centrale: al centro si osservano raramente guttae e la densità delle cellule endoteliali rimane normale. 1)
Iridodonesi: frequente negli occhi afachici, causa un trauma endoteliale intermittente.
Pigmentazione dell’angolo: la gonioscopia mostra un angolo ampio e profondo con vari gradi di pigmentazione del trabecolato.
Lo spessore centrale della cornea rimane normale (es. 541 μm), mentre la periferia edematosa (nasale e temporale) può presentare un ispessimento superiore a 700 μm. 1)
Un gonfiore transitorio della cornea centrale può essere osservato a causa dell’aumento della pressione intraoculare, ma l’edema centrale persistente è raro e limitato ai casi gravi.
Quando la sindrome di Brown-McLean si sviluppa nella cornea trapiantata, la pigmentazione e l’edema iniziano vicino al centro della cornea del donatore, ed è stato confermato che esiste una zona periferica priva di pigmento prima della giunzione tra innesto e ospite.
QLa sindrome di Brown-McLean può causare una significativa riduzione della vista?
A
Poiché la cornea centrale viene preservata, l’impatto sulla vista è spesso lieve. Tuttavia, se non trattata e progredisce fino alla scompenso della cornea centrale, può essere necessario un trapianto di cornea. Inoltre, la rottura di bolle epiteliali associate comporta il rischio di ulcera corneale infettiva.
La fisiopatologia della sindrome di Brown-McLean è ancora sconosciuta. Inizialmente si sospettava un’associazione con una distrofia endoteliale corneale sottostante, ma non è stata identificata alcuna distrofia o gene specifico come causa. La presenza di casi all’interno delle famiglie suggerisce anche una possibile predisposizione genetica. 2)
I principali fattori di rischio e le procedure chirurgiche associate sono i seguenti.
Occhio afachico dopo estrazione intracapsulare del cristallino: il contesto di insorgenza più classico e tipico
Altre chirurgie intraoculari: estrazione extracapsulare del cristallino, facoemulsificazione, cheratoplastica perforante, impianto di lente intraoculare in camera anteriore, ricostruzione del cristallino con vitrectomia
Fattori non post-operatori : sublussazione del cristallino, riassorbimento spontaneo del cristallino, endotelite corneale, cheratocono, glaucoma ad angolo chiuso, distrofia miotonica
Omocistinuria : Sono stati riportati casi di sindrome di Brown-McLean associata a sublussazione del cristallino o afachia correlata a malattie metaboliche2)
Iridodonesi (tremolio dell’iride) : si ritiene che provochi traumi endoteliali corneali intermittenti, favorendo l’insorgenza della malattia.
È stato anche dimostrato che il trauma endoteliale non è sempre necessario per lo sviluppo della sindrome di Brown-McLean, poiché esistono casi senza contatto irido-corneale all’esame con microscopia ultrasonica (UBM) e casi insorti dopo iridectomia. 2)
La diagnosi si basa sulla combinazione dei reperti clinici all’esame con lampada a fessura e dell’anamnesi. In pazienti con precedenti di chirurgia intraoculare come l’estrazione intracapsulare del cristallino o malattie non chirurgiche (ad esempio sublussazione del cristallino), se si osserva un edema corneale che si estende dalla periferia inferiore a tutta la circonferenza, si deve sospettare la sindrome di Brown-McLean.
Microscopia speculare (esame delle cellule endoteliali corneali) : Il numero e la morfologia delle cellule endoteliali della cornea centrale rimangono normali. L’endotelio della cornea periferica mostra spesso una diminuzione del numero di cellule o alterazioni morfologiche, ma può anche essere normale. L’acquisizione di un’immagine panoramica mediante microscopia speculare ad ampio campo conferma una distribuzione uniforme di cellule dense fino al confine tra le zone edematose e non edematose. 1)
Microscopia confocale in vivo : Nella cornea periferica si possono osservare ipertrofia dei nervi corneali, fibrosi dello strato di Bowman e forma e dimensioni irregolari dell’epitelio basale. L’endotelio della cornea centrale è generalmente normale e sono stati riportati casi di grandi cellule corneali e spessi nervi corneali nello stroma posteriore.
Tomografia a coerenza ottica del segmento anteriore (AS-OCT) : consente di valutare quantitativamente l’ispessimento corneale periferico. Ad esempio, è stata registrata una marcata differenza di oltre 700 μm in periferia rispetto a 541 μm al centro. 1)
Imaging Scheimpflug : consente di visualizzare contemporaneamente l’ispessimento corneale periferico e lo spessore normale centrale.
Microscopia elettronica : Nella cornea periferica della sindrome di Brown-McLean, si osserva uno strato di collagene posteriore anomalo nella membrana di Descemet e cellule endoteliali distrutte. La microscopia elettronica a scansione può mostrare una netta linea di demarcazione tra endotelio normale e patologico.
Esame gonioscopico: L’angolo è ampio e profondo, con vari gradi di pigmentazione del trabecolato. Negli occhi con edema corneale inferiore, è importante verificare anche la presenza di corpi estranei o residui di cristallino mediante gonioscopia.
QCome si differenzia la sindrome di Brown-McLean dalla distrofia endoteliale corneale di Fuchs?
A
Nella distrofia endoteliale di Fuchs, nella cornea centrale compaiono gocce corneali (guttae) e l’edema inizia dalla parte centrale, mentre nella sindrome di Brown-McLean le gocce corneali sono quasi assenti nella cornea centrale e l’edema inizia dalla parte periferica inferiore. Anche una storia di chirurgia intraoculare è un importante indizio per la diagnosi della sindrome di Brown-McLean.
La maggior parte dei casi di sindrome di Brown-McLean risponde al trattamento conservativo.
Collirio ipertonico salino : utilizzare collirio o unguento al cloruro di sodio al 5%. Mira alla riduzione osmotica dell’edema corneale. In un caso, l’uso di unguento NaCl 5% al momento di coricarsi ha migliorato l’acuità visiva corretta da 20/160 a 20/80 dopo 6 mesi. 2)
Collirio steroideo topico : utilizzato per ridurre l’edema e sopprimere l’infiammazione.
È stato anche dimostrato che l’uso di lenti a contatto può essere ben tollerato anche in presenza di edema corneale periferico.
Nei casi refrattari o con sintomi marcati, si considera l’intervento chirurgico.
Trapianto anulare di membrana amniotica (annular amniotic membrane transplant) : Per i casi ricorrenti di bolle epiteliali secondarie alla sindrome di Brown-McLean, il trapianto anulare di membrana amniotica, eseguito utilizzando due trapani di diametro diverso e posizionando il lato della membrana basale verso l’alto, è considerato efficace. Consente di trattare le bolle mantenendo la visione centrale.
Puntura stromale anteriore (anterior stromal puncture) : eseguita con ago 23G o 25G. Induce l’espressione di collagene, migliora l’adesione delle cellule epiteliali e la fibrosi subepiteliale, aumentando così la capacità di impedire l’ingresso di acqua nell’epitelio corneale.
Rimozione della lente intraoculare della camera anteriore: nella sindrome di Brown-McLean associata a una lente intraoculare della camera anteriore, la rimozione della lente spesso risolve l’edema corneale.
Trapianto di cornea : Nei casi in cui si verifichi uno scompenso corneale centrale non trattato, può essere necessario un trapianto di cornea.
6. Fisiopatologia e meccanismi dettagliati di insorgenza
Nella sindrome di Brown-McLean, le cellule endoteliali corneali (CEC) centrali mantengono un’alta densità mentre l’endotelio periferico è danneggiato. Di solito, nella distrofia endoteliale corneale di Fuchs, la diminuzione del numero e l’ipertrofia delle cellule endoteliali iniziano al centro e si estendono verso la periferia. 1) Al contrario, nella sindrome di Brown-McLean, l’endotelio periferico è selettivamente danneggiato mentre l’endotelio centrale denso rimane stabile a lungo termine, mostrando un pattern inverso.
In un caso di follow-up a lungo termine di 12 anni, le CEC centrali sono rimaste pressoché invariate da 2.499 cellule/mm² alla prima visita a 2.456 cellule/mm² dopo 12 anni, con un tasso di diminuzione annuale di solo lo 0,09 %. 1)
Per spiegare questa distribuzione peculiare sono state proposte le seguenti ipotesi. 1)
Formazione di una barriera biologica : Alla periferia si forma accidentalmente una struttura simile alla linea di Schwalbe (linea di confine corneale), che impedisce la migrazione cellulare dal centro alla periferia.
Selettività della direzione di migrazione delle cellule endoteliali : Le cellule endoteliali corneali preferiscono la migrazione centripeta (verso il centro) e hanno una scarsa capacità di migrazione centrifuga (verso la periferia).
Ostruzione della migrazione da parte di cellule in decomposizione : Le cellule endoteliali distrutte nell’area edematosa possono ostacolare fisicamente la migrazione cellulare dal centro alla periferia.
Si ipotizza che una predisposizione genetica alle malattie dell’endotelio corneale, combinata con un trauma all’endotelio, possa essere la causa della sindrome di Brown-McLean. L’iridodonesi (tremore dell’iride) causerebbe traumi endoteliali intermittenti, danneggiando selettivamente l’endotelio periferico.
L’assenza di contatto irido-corneale all’esame con microscopia ultrasonica biomicroscopica suggerisce che un trauma endoteliale non sia sempre necessario per lo sviluppo della sindrome di Brown-McLean.
Quando il danno all’endotelio corneale progredisce e le funzioni di pompa e barriera dell’endotelio scendono al di sotto di una soglia, il contenuto d’acqua dello stroma corneale aumenta e si verifica edema. Nella sindrome di Brown-McLean, questo danno è caratteristicamente limitato alla periferia.
QPerché la cornea centrale rimane trasparente a lungo?
A
Attualmente non esiste una risposta chiara. È stato proposto che si possa formare una barriera biologica (struttura simile alla linea di Schwalbe) al confine tra la zona centrale e quella periferica, o che le cellule endoteliali abbiano una scarsa capacità di migrazione centrifuga (dal centro alla periferia). 1) Risolvere questo mistero potrebbe portare allo sviluppo di nuovi trattamenti per le malattie dell’endotelio corneale.
7. Ricerche recenti e prospettive future (rapporti di fase di ricerca)
Un follow-up a lungo termine di 12 anni suggerisce che nella sindrome di Brown-McLean potrebbe formarsi una barriera biologica al confine tra centro e periferia. 1) Se questa ipotesi venisse confermata, si potrebbe prendere in considerazione un approccio terapeutico che preveda la creazione intenzionale di una barriera meccanica simile. Ciò potrebbe apportare una nuova prospettiva per la comprensione e lo sviluppo di trattamenti per le malattie dell’endotelio corneale in generale.
Tomioka et al. (2024) hanno riportato un caso di follow-up a lungo termine di 12 anni presso la Kyoto Prefectural University of Medicine. 1) Le immagini panoramiche ottenute mediante microscopia speculare a contatto a scansione di fessura a campo largo hanno mostrato che CEC ad alta densità, equivalenti a quelle della regione centrale, erano uniformemente distribuite fino a poco prima del confine tra zona edematosa e non edematosa. Un tasso di diminuzione annuale delle CEC centrali dello 0,09%, estremamente stabile, suggerisce l’esistenza di una dinamica endoteliale unica nella sindrome di Brown-McLean.
Riconoscimento dei casi associati a malattie sistemiche come l’omocistinuria
È stato riportato che in pazienti con malattie metaboliche come l’omocistinuria, la sindrome di Brown-McLean può insorgere dopo sublussazione del cristallino o afachia. 2) L’istituzione di un protocollo di diagnosi e follow-up per questi casi con malattie sistemiche associate è considerata una sfida futura.
Alenezi et al. (2021) hanno riportato il primo caso di sindrome di Brown-McLean associata a omocistinuria in Medio Oriente. 2) Un uomo di 29 anni ha sviluppato la sindrome all’occhio sinistro più di 22 anni dopo vitrectomia e lensectomia per sublussazione bilaterale del cristallino. Un trattamento conservativo con unguento al 5% di NaCl ha migliorato l’acuità visiva corretta da 20/160 a 20/80. Inoltre, la presenza di un’ampia iridectomia inferiore non ha impedito lo sviluppo di edema periferico inferiore, un’osservazione importante che confuta l’ipotesi tradizionale del ruolo protettivo dell’iridectomia superiore.
Tomioka Y, Tanaka H, Sotozono C, Kinoshita S. A comprehensive long-term follow-up study of Brown-McLean syndrome. American journal of ophthalmology case reports. 2024;36:102146. doi:10.1016/j.ajoc.2024.102146. PMID:39282598; PMCID:PMC11393604.
Alenezi SH, Alrefaie SM, Alreshidi SO, ALBalawi HB, Osorio HM.. Brown-Mclean syndrome in an aphakic patient with homocystinuria: The first reported case in Middle East. Saudi J Ophthalmol. 2020;34(4):300-302. doi:10.4103/1319-4534.322619. PMID:34527877; PMCID:PMC8409359.
Chatterjee S, Parchand SM, Dash D, Agrawal D. Brown-McLean syndrome revisited. Indian J Ophthalmol. 2020;68(1):183-184. PMID: 31856505.
Copia il testo dell'articolo e incollalo nell'assistente IA che preferisci.
Articolo copiato negli appunti
Apri un assistente IA qui sotto e incolla il testo copiato nella chat.