La retinopatía cristalina es un término general para un grupo heterogéneo de enfermedades caracterizadas por depósitos de cristales en cualquier capa o región de la retina. Las causas son diversas, incluyendo enfermedades hereditarias, efectos secundarios de medicamentos y complicaciones de enfermedades sistémicas.
La clasificación de las causas es la siguiente:
Hereditarias: Distrofia retiniana cristalina de Bietti (BCD), cistinosis, hiperoxaluria primaria, síndrome de Sjögren-Larsson
Las retinopatías cristalinas hereditarias representativas incluyen las siguientes:
Distrofia retiniana cristalina de Bietti (BCD): Un trastorno autosómico recesivo causado por mutaciones en el gen CYP4V2 (4q35), reportado por primera vez por Bietti en 1937. Las tres características principales son manchas blanco-amarillentas cristalinas brillantes dispersas en el polo posterior, atrofia de la coriocapilar y depósitos cristalinos en la córnea, aunque se han reportado muchos casos sin afectación corneal. Es más común en Asia oriental, especialmente en poblaciones japonesa y china. Representa el 10% de los casos diagnosticados como retinitis pigmentosa no sindrómica autosómica recesiva.
Cistinosis: Enfermedad de almacenamiento lisosomal autosómica recesiva causada por mutaciones en el gen CTNS (17p13.2).
Hiperoxaluria primaria (PH): Trastorno autosómico recesivo causado por mutaciones en los genes AGXT/GRHPR/HOGA1.
Síndrome de Sjögren-Larsson: Trastorno autosómico recesivo causado por mutaciones en el gen ALDH3A2 (17p11.2).
La retinopatía cristalina se considera una enfermedad relacionada con la retinitis pigmentosa. 1)
Q¿Qué es la distrofia cristalina de Bietti (BCD)?
A
Es una enfermedad coriorretiniana autosómica recesiva causada por mutaciones en el gen CYP4V2, caracterizada por depósitos cristalinos en el polo posterior, atrofia del EPR y la coroides, y cristales corneales. Es más común en poblaciones de Asia oriental, presentándose con ceguera nocturna y defectos del campo visual en los 20 a 40 años, y llevando a discapacidad visual severa en los 50 a 60 años. Actualmente no existe un tratamiento curativo, pero se están realizando investigaciones sobre terapia génica.
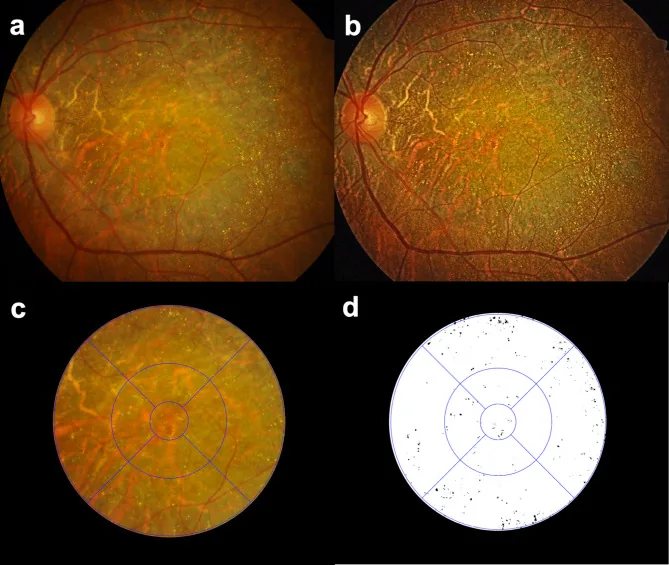
Seung Jun You; Chang Ki Yoon; Un Chul Park; Kyu Hyung Park; Eun Kyoung Lee. Longitudinal quantitative assessment of retinal crystalline deposits in bietti crystalline dystrophy. BMC Ophthalmol. 2025 Mar 17; 25:139. Figure 1. PMCID: PMC11916969. License: CC BY.
Evaluación cuantitativa de depósitos cristalinos retinianos en ojos con distrofia cristalina de Bietti. (a) Fotografía de fondo de ojo a color que muestra atrofia coriorretiniana grisácea y numerosos depósitos cristalinos amarillo-blancos. (b) Contraste de imagen mejorado mediante ecualización de histograma adaptativa limitada por contraste (CLAHE) para detectar mejor los depósitos cristalinos retinianos. (c) Cuadrícula del Estudio de Tratamiento Temprano de la Retinopatía Diabética (ETDRS) superpuesta para la cuantificación regional de depósitos cristalinos retinianos. (d) Cristales retinianos extraídos como negro sobre fondo blanco utilizando el software Medilabel®
Los síntomas subjetivos de cada enfermedad son los siguientes. Se presentan con pérdida progresiva del campo visual y de la agudeza visual, pero el pronóstico varía según el caso. 1)
BCD:
Ceguera nocturna progresiva y defectos del campo visual (escotomas paracentrales) que se desarrollan en los 20 a 40 años.
Posteriormente, se produce una pérdida rápida de la agudeza visual, que lleva a discapacidad visual severa en los 50 a 60 años.
Los hallazgos clínicos de BCD se organizan según la clasificación de tres etapas de Yuzawa.
A continuación se muestra la correspondencia de la deposición de cristales y la atrofia del EPR en cada etapa.
Estadio
Depósitos cristalinos
Atrofia del EPR
1
Numerosos en el polo posterior
Leve, macular
2
Disminuidos en el polo posterior
Progresiva, extensa
3
Casi ausentes
Grave, difusa
Estadio 1: Numerosos cristales pequeños, brillantes, de color blanco amarillento, dispersos desde el polo posterior hasta la periferia media. Se encuentran a nivel del complejo EPR-coriocapilar. Se asocia con atrofia leve del EPR macular.
Estadio 2: Atrofia progresiva del EPR y atrofia coriorretiniana que se extiende más allá del polo posterior. Los cristales disminuyen en el polo posterior y permanecen en la periferia media.
Estadio 3: Atrofia extensa del EPR y de la coriocapilar. Los cristales están casi ausentes.
Algunos casos presentan cristales en el estroma corneal en la periferia anterior de la córnea, pero no están presentes en todos los casos.
Cristales de oxalato amarillos: distribuidos principalmente en la capa plexiforme externa y nuclear externa alrededor de la fóvea y a lo largo de las arterias
Lesiones anulares subretinianas negras por proliferación del EPR, que progresan a atrofia geográfica
También se evalúa mediante la clasificación de Derveaux (grados 1 a 4).
Grado 1: Cristales de oxalato perifoveales aislados
Grado 2: Cristales maculares que respetan la fóvea + proliferación del EPR
Grado 3: Proliferación del EPR, fibrosis subretiniana y fibrosis foveal
BCD (CYP4V2): Autosómico recesivo. Común en asiáticos orientales. Anomalías en enzimas del metabolismo de lípidos/esteroides. 1)
Cistinosis (CTNS): Autosómico recesivo. Ocurre en 1 de cada 100,000–200,000 nacimientos. Enfermedad de almacenamiento lisosomal.
Hiperoxaluria (AGXT, etc.): Autosómico recesivo. Prevalencia menor de 3 por 1,000,000.
Síndrome de Sjögren–Larsson (ALDH3A2): Autosómico recesivo. Metabolismo anormal de aldehídos grasos.
Inducido por fármacos
Tamoxifeno: Depósitos cristalinos en la capa de fibras nerviosas y la capa plexiforme interna.
Cantaxantina: Depósitos por colorante alimentario (suplementos).
Talco: Excipiente de comprimidos. Puede causar retinopatía por talco tras administración intravenosa.
Metoxiflurano y nitrofurantoína: Uso actualmente limitado.
Otros
Degeneración: Depósitos cristalinos asociados con cambios degenerativos crónicos de la retina.
Idiopático: Depósitos cristalinos de causa desconocida.
Iatrogénico: Depósito resultante de un tratamiento médico.
Q¿Cuáles son las causas de la retinopatía cristalina?
A
Las causas principales son hereditarias (BCD, cistinosis, hiperoxaluria, síndrome de Sjögren-Larsson) y farmacológicas (tamoxifeno, cantaxantina, talco, etc.). Dado que el tratamiento y el pronóstico varían mucho según la causa, el diagnóstico preciso de la causa es esencial para determinar la estrategia de tratamiento.
Además de los hallazgos característicos del fondo de ojo, son útiles la electrorretinografía, la FA, la ICGA y la OCT.
FA e ICGA
FA: Se observa hiperfluencia por defectos en ventana en áreas de atrofia del EPR.
ICGA: Retraso en el llenado coroideo en todas las etapas. En fase tardía muestra hipofluencia parcheada.
OCT y FAF
SD-OCT: Puntos hiperreflectivos en todas las capas retinianas (principalmente en el complejo RPE-membrana de Bruch). Pueden observarse tubulaciones retinianas externas (ORTs) en la capa nuclear externa.
FAF: Los cristales no son visibles por sí mismos. Las células RPE dañadas muestran hiperautofluorescencia granular, mientras que las áreas de atrofia del RPE muestran hipoautofluorescencia. La luz infrarroja cercana (NIR) visualiza bien los cristales.
Electrorretinografía
Evaluación funcional: Normal → disminuida → ausente según el estadio. La disfunción de patrón cono-bastón es común.
Aplicación en diagnóstico diferencial: En BCD no hay estrechamiento de los vasos retinianos y la respuesta electrorretinográfica está relativamente preservada, lo que ayuda a diferenciarla de la retinitis pigmentosa. Las pruebas genéticas también son útiles.
Cistinosis: La medición de la concentración de cistina libre no unida a proteínas en leucocitos polimorfonucleares es útil para el diagnóstico definitivo.
Hiperoxaluria primaria: Aumento de la excreción de oxalato (más del doble de lo normal) en orina de 24 horas. Las pruebas genéticas (AGXT/GRHPR/HOGA1) también pueden confirmar.
Síndrome de Sjögren-Larsson: Pruebas genéticas (ALDH3A2) o medición de la actividad de FALDH (aldehído graso deshidrogenasa).
Retinopatía por tamoxifeno: En el diagnóstico diferencial considerar retinopatía cristalina por CYP4V2, drusas y telangiectasia parafoveal.
Q¿Cómo se distingue la retinopatía cristalina de la retinitis pigmentosa?
A
En BCD, no hay estrechamiento de los vasos retinianos y la respuesta electrorretinográfica está relativamente preservada. Las pruebas genéticas (CYP4V2) son útiles para el diagnóstico definitivo, y aproximadamente el 10% de los casos diagnosticados como retinitis pigmentosa no sindrómica autosómica recesiva son en realidad BCD.
Actualmente no existe un tratamiento curativo eficaz. El cuidado de la baja visión es fundamental, y se realiza el siguiente manejo para las complicaciones.
La suspensión del tamoxifeno detiene la progresión y, en ocasiones, produce mejoría.
Los fármacos anti-VEGF pueden ser eficaces para el edema macular quístico.
Q¿Se pueden curar los depósitos cristalinos retinianos causados por el tamoxifeno?
A
La suspensión del fármaco detiene la progresión y, a veces, mejora. Si se acompaña de edema macular quístico (CME), los agentes anti-VEGF pueden ser efectivos. En cualquier caso, la detección temprana es clave para mejorar el pronóstico.
Los mecanismos de cada enfermedad son los siguientes.
BCD: CYP4V2 codifica una enzima de la familia del citocromo P450 involucrada en el metabolismo de lípidos y esteroides. Las mutaciones de CYP4V2 causan que metabolitos lipídicos anormales se depositen en la coriorretina, lo que lleva a atrofia del EPR y la coriocapilar, y degeneración secundaria de los fotorreceptores.
Cistinosis: La disfunción de cistinosina (transportador de cistina de la membrana lisosomal) impide el transporte de cistina fuera de los lisosomas. La acumulación de cistina en las células endoteliales de la retina causa degeneración retiniana.
Hiperoxaluria primaria: Las anomalías congénitas en el metabolismo del glioxilato hepático conducen a una sobreproducción de oxalato y glicolato. Como resultado, los cristales de oxalato de calcio se depositan en la retina, el EPR y la coroides, provocando una degeneración retiniana progresiva.
Síndrome de Sjögren-Larsson: La deficiencia de FALDH (aldehído deshidrogenasa graso) conduce a la acumulación de aldehídos y alcoholes grasos, causando daño a las células de Müller y los fotorreceptores.
Retinopatía por tamoxifeno: El tamoxifeno se une a los lípidos y se acumula en los lisosomas, reduciendo la actividad enzimática y provocando depósitos cristalinos en la capa de fibras nerviosas y la capa plexiforme interna.
Se está llevando a cabo un ensayo clínico de fase 1 de rAAV2/8-hCYP4V2 (administración subretiniana) como terapia de reemplazo génico para BCD. Las pruebas genéticas tempranas y el asesoramiento genético pueden permitir que los pacientes se beneficien de la terapia génica en el futuro.