La rétinopathie cristalline est un terme générique désignant un groupe hétérogène de maladies caractérisées par des dépôts cristallins dans n’importe quelle couche ou région de la rétine. Les causes sont variées : maladies héréditaires, effets secondaires de médicaments, complications de maladies systémiques, etc.
Les principales rétinopathies cristallines héréditaires sont les suivantes :
Dystrophie cristalline de Bietti (BCD) : maladie autosomique récessive due à des mutations du gène CYP4V2 (4q35), décrite par Bietti en 1937. Les trois caractéristiques principales sont des dépôts cristallins scintillants jaune-blanc disséminés dans le pôle postérieur, une atrophie de la choriocapillaire et des dépôts cristallins cornéens, mais de nombreux cas sans atteinte cornéenne ont été rapportés. Fréquente en Asie de l’Est, notamment chez les Japonais et les Chinois. Environ 10 % des cas diagnostiqués comme RP non syndromique autosomique récessive seraient en fait une BCD.
Cystinose : Maladie de surcharge lysosomale autosomique récessive due à des mutations du gène CTNS (17p13.2).
Hyperoxalurie primitive (PH) : Maladie autosomique récessive due à des mutations des gènes AGXT/GRHPR/HOGA1.
Syndrome de Sjögren-Larsson : Maladie autosomique récessive due à des mutations du gène ALDH3A2 (17p11.2).
La rétinopathie cristalline est considérée comme une maladie apparentée à la rétinite pigmentaire. 1)
QQu'est-ce que la dystrophie cristalline de Bietti (BCD) ?
A
Maladie choroïdorétinienne autosomique récessive due à des mutations du gène CYP4V2, caractérisée par des dépôts cristallins au pôle postérieur, une atrophie de l’EPR et de la choroïde, et des cristaux cornéens. Plus fréquente chez les populations d’Asie de l’Est, elle se manifeste par une héméralopie et des troubles du champ visuel entre 20 et 40 ans, et conduit à une déficience visuelle sévère entre 50 et 60 ans. Il n’existe actuellement aucun traitement curatif, mais des recherches sur la thérapie génique sont en cours.
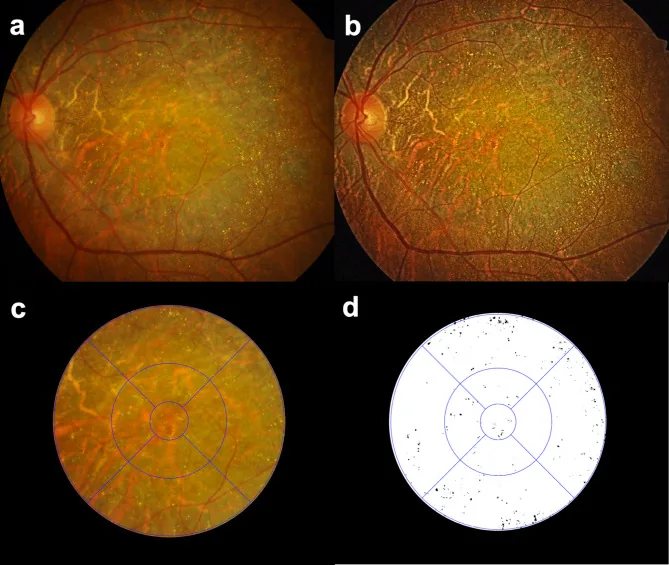
Seung Jun You; Chang Ki Yoon; Un Chul Park; Kyu Hyung Park; Eun Kyoung Lee. Longitudinal quantitative assessment of retinal crystalline deposits in bietti crystalline dystrophy. BMC Ophthalmol. 2025 Mar 17; 25:139. Figure 1. PMCID: PMC11916969. License: CC BY.
Évaluation quantitative des dépôts cristallins rétiniens dans les yeux atteints de dystrophie cristalline de Bietti. (a) Photographie couleur du fond d’œil montrant une atrophie choroïdorétinienne grisâtre et de nombreux dépôts cristallins jaune-blanc. (b) Contraste de l’image amélioré par égalisation d’histogramme adaptative à contraste limité (CLAHE) pour mieux détecter les dépôts cristallins rétiniens. (c) Grille ETDRS superposée pour la quantification régionale des dépôts cristallins rétiniens. (d) Cristaux rétiniens extraits en noir sur fond blanc à l’aide du logiciel Medilabel®.
Les symptômes subjectifs de chaque maladie sont les suivants. Ils présentent des troubles progressifs du champ visuel et de l’acuité visuelle, mais le pronostic varie selon les cas. 1)
BCD :
Héméralopie progressive et troubles du champ visuel (scotome paracentral) apparaissant entre 20 et 40 ans.
Par la suite, baisse rapide de l’acuité visuelle, conduisant à une déficience visuelle sévère entre 50 et 60 ans.
Photophobie et blépharospasme dus aux dépôts de cystine dans la cornée et la conjonctive au cours de la première année de vie
Baisse de l’acuité visuelle et rétrécissement du champ visuel dus à une dégénérescence rétinienne progressive
La cystinose néphropathique infantile (95 %) se manifeste par un retard de croissance et une acidose tubulaire rénale, conduisant à une insuffisance rénale à l’adolescence
Hyperoxalurie primitive :
Lithiase rénale récurrente et coliques néphrétiques apparaissant chez 50 % des patients avant l’âge de 5 ans
La baisse de l’acuité visuelle est plus probablement due à une atrophie optique qu’à des dépôts cristallins rétiniens
90 % des patients présentent des symptômes avant l’âge de 25 ans
Les signes cliniques de la BCD sont classés selon la classification en 3 stades de Yuzawa.
Le tableau ci-dessous montre la correspondance entre les dépôts cristallins et l’atrophie de l’EPR à chaque stade.
Stade
Dépôts cristallins
Atrophie de l’EPR
1
Nombreux au pôle postérieur
Légère, maculaire
2
Diminués au pôle postérieur
Progressive, étendue
3
Presque disparus
Sévère, généralisée
Stade 1 : Nombreux petits cristaux jaune-blanc brillants dispersés du pôle postérieur à la périphérie moyenne. Ils se situent au niveau du complexe EPR-choriocapillaire. Atrophie légère de l’EPR maculaire associée.
Stade 2 : Atrophie progressive de l’EPR et de la choroïde/rétine s’étendant au-delà du pôle postérieur. Les cristaux diminuent au pôle postérieur et persistent dans la périphérie moyenne.
Stade 3 : Atrophie étendue de l’EPR et de la choriocapillaire. Les cristaux ont presque disparu.
Certains cas présentent des cristaux dans le stroma cornéen antérieur près du limbe, mais ils ne sont pas présents dans tous les cas.
Cristaux d’oxalate jaunes : principalement distribués autour de la fovéa, dans la couche plexiforme externe et la couche nucléaire externe autour des artères
Lésions sous-rétiniennes annulaires noires dues à la prolifération de l’EPR, évoluant vers une atrophie géographique
Elle est également évaluée par le grading de Derveaux (grades 1 à 4).
Grade 1 : cristaux d’oxalate isolés autour de la fovéa
Grade 2 : cristaux maculaires épargnant la fovéa + prolifération de l’EPR
Grade 3 : prolifération de l’EPR, fibrose sous-rétinienne et fibrose fovéolaire
BCD (CYP4V2) : autosomique récessif. Fréquent chez les populations d’Asie de l’Est. Anomalie des enzymes du métabolisme des lipides et des stéroïdes. 1)
Cystinose (CTNS) : autosomique récessif. 1 naissance sur 100 000 à 200 000. Maladie de surcharge lysosomale.
Hyperoxalurie (AGXT etc.) : autosomique récessif. Prévalence inférieure à 3 personnes par million.
Syndrome de Sjögren-Larsson (ALDH3A2) : autosomique récessif. Anomalie du métabolisme des aldéhydes aliphatiques.
Médicamenteux
Tamoxifène : dépôts cristallins dans la couche des fibres nerveuses et la couche plexiforme interne.
Canthaxanthine : dépôts dus au colorant alimentaire (suppléments).
Talc : excipient de comprimés. Peut provoquer une rétinopathie talcique après administration intraveineuse.
Dégénérescence : dépôts cristallins associés à des modifications dégénératives chroniques de la rétine.
Idiopathique : dépôts cristallins de cause inconnue.
Iatrogène : dépôt résultant d’un acte thérapeutique.
QQuelles sont les causes de la rétinopathie cristalline ?
A
Les causes principales sont héréditaires (BCD, cystinose, hyperoxalurie, syndrome de Sjögren-Larsson) et médicamenteuses (tamoxifène, canthaxanthine, talc, etc.). Le traitement et le pronostic varient considérablement selon la cause, donc un diagnostic précis est essentiel pour déterminer la stratégie thérapeutique.
En plus des signes caractéristiques du fond d’œil, l’électrorétinographie, l’angiographie à la fluorescéine (FA), l’angiographie au vert d’indocyanine (ICGA) et l’OCT sont utiles.
FA et ICGA
FA : On observe une hyperfluorescence due à des fenêtres de transmission dans les zones d’atrophie de l’EPR.
ICGA : Retard de remplissage choroïdien à tous les stades. Hypofluorescence en forme de plaque aux stades tardifs.
OCT et FAF
SD-OCT : Points hyperréflectifs dans toutes les couches rétiniennes (principalement au niveau du complexe RPE-membrane de Bruch). Des structures tubulaires rétiniennes externes (ORTs) peuvent être observées dans la couche nucléaire externe.
FAF : Les cristaux eux-mêmes sont invisibles. Les cellules RPE endommagées présentent une autofluorescence granuleuse intense, tandis que les zones d’atrophie RPE montrent une hypo-autofluorescence. La lumière proche infrarouge (NIR) visualise bien les cristaux.
Électrorétinogramme
Évaluation fonctionnelle : Normale → diminuée → absente selon le stade. Un dysfonctionnement de type cône-bâtonnet est courant.
Application au diagnostic différentiel : Dans la BCD, il n’y a pas de rétrécissement des vaisseaux rétiniens et la réponse électrorétinographique est relativement préservée, ce qui aide à la différencier de la rétinite pigmentaire. Les tests génétiques sont également utiles.
Cystinose : La mesure de la concentration de cystine libre non liée aux protéines dans les leucocytes polymorphonucléaires est utile pour le diagnostic définitif.
Hyperoxalurie primitive : Confirmation d’une excrétion accrue d’oxalate (au moins le double de la normale) dans les urines de 24 heures. Les tests génétiques (AGXT/GRHPR/HOGA1) peuvent également confirmer le diagnostic.
Syndrome de Sjögren-Larsson : Tests génétiques (ALDH3A2) ou mesure de l’activité FALDH (aldéhyde déshydrogénase aliphatique).
Rétinopathie au tamoxifène : Le diagnostic différentiel inclut la rétinopathie cristalline CYP4V2, les drusen et la télangiectasie périfovéolaire.
QComment distinguer la rétinopathie cristalline de la rétinite pigmentaire ?
A
Dans la BCD, il n’y a pas de rétrécissement des vaisseaux rétiniens et la réponse électrorétinographique est relativement préservée. Les tests génétiques (CYP4V2) sont utiles pour le diagnostic définitif, et environ 10 % des cas diagnostiqués comme rétinite pigmentaire non syndromique autosomique récessive sont en fait une BCD.
Il n’existe actuellement aucun traitement curatif efficace. Les soins de basse vision sont essentiels, et la gestion des complications est la suivante :
L’arrêt du traitement stoppe la progression et peut parfois entraîner une amélioration.
Les anti-VEGF peuvent être efficaces en cas d’œdème maculaire cystoïde.
QLes dépôts cristallins rétiniens dus au tamoxifène sont-ils réversibles ?
A
L’arrêt du traitement stoppe la progression et permet parfois une amélioration. En cas d’œdème maculaire cystoïde (OMC) associé, les anti-VEGF peuvent être efficaces. Dans tous les cas, la détection précoce est la clé pour améliorer le pronostic.
Les mécanismes de chaque maladie sont les suivants :
BCD : CYP4V2 code une enzyme de la famille du cytochrome P450, impliquée dans le métabolisme des lipides et des stéroïdes. Les mutations de CYP4V2 entraînent le dépôt de métabolites lipidiques anormaux dans la choroïde et la rétine, conduisant à l’atrophie de l’EPR et de la choriocapillaire, puis à une dégénérescence secondaire des photorécepteurs.
Cystinose : Le dysfonctionnement de la cystinosine (protéine de transport de la cystine dans la membrane lysosomale) inhibe le transport de la cystine hors des lysosomes. L’accumulation de cystine dans les cellules endothéliales rétiniennes provoque une dégénérescence rétinienne.
Hyperoxalurie primitive : Une anomalie congénitale du métabolisme du glyoxylate dans le foie entraîne une surproduction d’oxalate et de glycolate. En conséquence, des cristaux d’oxalate de calcium se déposent dans la rétine, l’EPR et la choroïde, provoquant une dégénérescence rétinienne progressive.
Syndrome de Sjögren-Larsson : Le déficit en FALDH (aldéhyde déshydrogénase aliphatique) entraîne l’accumulation d’aldéhydes et d’alcools aliphatiques, endommageant les cellules de Müller et les photorécepteurs.
Rétinopathie au tamoxifène : Le tamoxifène se lie aux lipides et s’accumule dans les lysosomes, réduisant l’activité enzymatique et provoquant le dépôt de substances cristallines dans la couche des fibres nerveuses et la couche plexiforme interne.
Un essai clinique de phase 1 est en cours pour la thérapie génique de substitution de BCD, utilisant le rAAV2/8-hCYP4V2 (injection sous-rétinienne). Un dépistage génétique précoce et un conseil génétique pourraient permettre de bénéficier de futures thérapies géniques.