Kristalin retinopati (Crystalline retinopathy), retinanın herhangi bir tabakasında veya bölgesinde kristal birikintileri görülen heterojen bir hastalık grubunun genel adıdır. Nedenler kalıtsal hastalıklar, ilaç yan etkileri ve sistemik hastalık komplikasyonlarını içerir.
Yaygın kalıtsal kristalin retinopatiler şunlardır:
Bietti kristalin retinal distrofi (BCD): CYP4V2 gen mutasyonuna (4q35) bağlı otozomal resesif bir hastalık. İlk kez 1937’de Bietti tarafından tanımlanmıştır. Üç ana özelliği arka kutupta dağınık parlak kristalimsi sarı-beyaz lekeler, koroid kapiller atrofisi ve korneada kristal birikintileridir, ancak kornea tutulumu olmayan birçok vaka bildirilmiştir. Doğu Asya’da, özellikle Japon ve Çinlilerde yaygındır. Otozomal resesif nonsendromik RP tanısı alan vakaların yaklaşık %10’u aslında BCD’dir.
Sistinoz: CTNS gen mutasyonuna (17p13.2) bağlı otozomal resesif geçişli bir lizozomal depo hastalığı.
Primer hiperoksalüri (PH): AGXT/GRHPR/HOGA1 gen mutasyonlarına bağlı otozomal resesif geçişli bir hastalık.
Sjögren-Larsson sendromu: ALDH3A2 gen mutasyonuna (17p11.2) bağlı otozomal resesif geçişli bir hastalık.
Kristalin retinopati, retinitis pigmentozanın ilişkili bir hastalığı olarak kabul edilir. 1)
QBietti Kristalin Retinal Distrofisi (BCD) nedir?
A
CYP4V2 gen mutasyonuna bağlı otozomal resesif geçişli bir koroid-retina hastalığıdır; arka kutupta kristal birikimi, RPE ve koroid atrofisi ve kornea kristalleri ile karakterizedir. Doğu Asya popülasyonunda daha sık görülür, 20-40 yaşlarında gece körlüğü ve görme alanı defekti ile başlar ve 50-60 yaşlarında ileri görme kaybına yol açar. Şu anda kesin bir tedavisi yoktur ve gen tedavisi araştırmaları devam etmektedir.
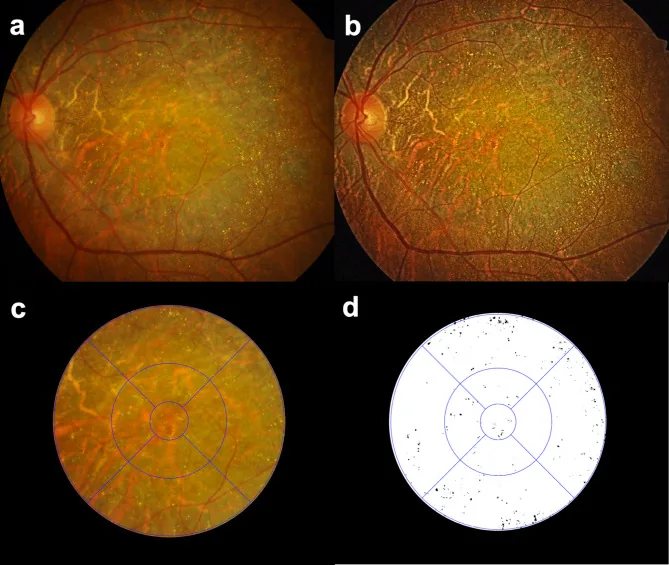
Seung Jun You; Chang Ki Yoon; Un Chul Park; Kyu Hyung Park; Eun Kyoung Lee. Longitudinal quantitative assessment of retinal crystalline deposits in bietti crystalline dystrophy. BMC Ophthalmol. 2025 Mar 17; 25:139. Figure 1. PMCID: PMC11916969. License: CC BY.
Bietti Kristalin Distrofisi olan gözlerde retinal kristal birikintilerinin kantitatif değerlendirmesi. (a) Gri renkli koroid-retina atrofisi ve çok sayıda sarı-beyaz kristal birikintisini gösteren renkli fundus fotoğrafı. (b) Retinal kristal birikintilerini daha iyi tespit etmek için Contrast Limited Adaptive Histogram Equalization (CLAHE) kullanılarak kontrastı artırılmış görüntü. (c) Retinal kristal birikintilerinin bölgesel kantifikasyonu için ETDRS ızgarası yerleştirilmiş. (d) Medilabel® yazılımı kullanılarak beyaz zemin üzerinde siyah olarak çıkarılmış retinal kristaller.
Her hastalığın sübjektif belirtileri aşağıdaki gibidir. Progresif görme alanı defekti ve görme azalması ile seyreder, ancak prognoz vakalar arasında değişkendir. 1)
BCD:
20-40 yaşlarında progresif gece körlüğü ve görme alanı defekti (parasantral skotom) başlar.
Ardından hızlı görme kaybı gelişir ve 50-60 yaşlarında ileri görme kaybı oluşur.
BCD’nin klinik bulguları Yuzawa’nın 3 evreli sınıflamasına göre düzenlenir.
Aşağıda her evredeki kristal birikimi ve RPE atrofisi gösterilmiştir.
Evre
Kristal birikimi
RPE atrofisi
1
Arka kutupta çok sayıda
Hafif, makula bölgesi
2
Arka kutupta azalma
İlerleyici, yaygın
3
Neredeyse kaybolmuş
Şiddetli, tüm alan
Evre 1: Arka kutuptan orta perifere kadar çok sayıda küçük, parlak sarı-beyaz kristal dağılmıştır. RPE-koryokapillaris kompleksi seviyesinde bulunurlar. Hafif makulaRPE atrofisi eşlik eder.
Evre 2: Arka kutup ötesine ilerleyici RPE atrofisi ve koryoretinal atrofi. Kristaller arka kutupta azalır ve orta periferde kalır.
Evre 3: Yaygın RPE ve koryokapillaris atrofisi. Kristaller neredeyse kaybolmuştur.
Bazı olgularda kornea limbusunun önündeki korneal stromada kristaller görülür, ancak tüm olgularda mevcut değildir.
Talk: Tablet dolgu maddesi. İntravenöz uygulama sonrası talk retinopatisine neden olabilir.
Metoksifluran ve Nitrofurantoin: Günümüzde kullanımı sınırlıdır.
Diğer
Dejeneratif: Retinanın kronik dejeneratif değişikliklerine eşlik eden kristal benzeri birikim.
İdiyopatik: Nedeni bilinmeyen kristal birikimi.
İyatrojenik: Tedaviye bağlı birikim.
QKristalin retinopatinin nedenleri nelerdir?
A
Başlıca nedenler kalıtsal (BCD, sistinoz, hiperoksalüri, Sjögren-Larsson sendromu) ve ilaca bağlı (tamoksifen, kantaksantin, talk vb.) olarak sıralanabilir. Tedavi ve prognoz nedene göre büyük farklılık gösterdiğinden, tedavi planının belirlenmesinde doğru neden teşhisi esastır.
FA: RPE atrofi alanlarında pencere defektine bağlı hiperfloresans görülür.
ICGA: Tüm evrelerde koroidal dolum gecikmesi. Geç dönemde plak benzeri hipofloresans gösterir.
OCT ve FAF
SD-OCT: Tüm retina katmanlarında yüksek yansıtıcı noktalar (çoğunlukla RPE-Bruch membran kompleksinde). Dış nükleer tabakada dış retina tübüler yapıları (ORT’ler) görülebilir.
FAF: Kristallerin kendisi görünmez. Hasar görmüş RPE hücreleri granüler yoğun otofloresans gösterir, RPE atrofisi alanlarında ise düşük otofloresans görülür. Yakın kızılötesi ışık (NIR) kristalleri iyi gösterir.
Elektroretinografi
Fonksiyonel değerlendirme: Evreye göre normal → azalmış → kaybolmuş. Koni-baston paterninde işlev bozukluğu yaygındır.
Ayırıcı tanıda kullanım: BCD’de retina damarlarında daralma olmaz ve elektroretinografi yanıtı nispeten korunur, bu da retinitis pigmentozadan ayırmada yardımcıdır. Genetik test de faydalıdır.
Sistinoz: Polimorfonükleer lökositlerde serbest protein bağlı olmayan sistin konsantrasyonunun ölçümü kesin tanıda faydalıdır.
Primer hiperoksalüri: 24 saatlik idrarda oksalat atılımında artış (normalin iki katından fazla) doğrulanır. Genetik test (AGXT/GRHPR/HOGA1) de kesin tanı koyabilir.
Sjögren-Larsson sendromu: Genetik test (ALDH3A2) veya FALDH (yağlı aldehit dehidrogenaz) aktivite ölçümü.
QKristalin retinopati retinitis pigmentozadan nasıl ayırt edilir?
A
BCD’de retina damarlarında daralma olmaz ve elektroretinografi yanıtı nispeten korunur. Genetik test (CYP4V2) kesin tanıda faydalıdır ve otozomal resesif nonsendromik retinitis pigmentoza tanısı alan vakaların yaklaşık %10’u aslında BCD’dir.
İlacın kesilmesi ilerlemeyi durdurur ve bazen düzelme sağlar.
Kistoid makula ödeminde anti-VEGF ilaçlar etkili olabilir.
QTamoksifene bağlı retina kristal birikintileri iyileşir mi?
A
İlacın kesilmesiyle ilerleme durur ve bazen gerileme görülür. Kistoid makula ödemi (CME) eşlik ediyorsa anti-VEGF ilaçlar etkili olabilir. Her iki durumda da erken teşhis, prognozun iyileşmesinde anahtardır.
Her hastalığın oluşum mekanizması aşağıdaki gibidir:
BCD: CYP4V2, sitokrom P450 ailesinden bir enzimi kodlar ve lipid/steroid metabolizmasında rol oynar. CYP4V2 mutasyonu, anormal lipid metabolitlerinin retina ve koroidde birikmesine, RPE ve koroid kapiller tabakasının atrofisine ve ikincil olarak fotoreseptör dejenerasyonuna yol açar.
Primer hiperoksalüri: Karaciğerde glikoksilat metabolizmasının konjenital anomalisi nedeniyle aşırı oksalat ve glikolat üretimi olur. Sonuçta kalsiyum oksalat kristalleri retina, RPE ve koroidde birikir ve retina dejenerasyonu ilerler.
Sjögren-Larsson sendromu: FALDH (yağlı aldehit dehidrogenaz) eksikliği, yağlı aldehit ve alkollerin birikmesine yol açarak Müller hücrelerine ve fotoreseptörlere hasar verir.
Tamoksifen retinopatisi: Tamoksifen lipidlerle birleşerek lizozomlarda birikir ve enzim aktivitesini azaltarak sinir lifi tabakası ve iç pleksiform tabakada kristal benzeri maddelerin birikmesine neden olur.
BCD için gen replasman tedavisi olarak rAAV2/8-hCYP4V2 (subretinal enjeksiyon) faz 1 klinik çalışması devam etmektedir. Erken genetik test ve genetik danışmanlık sayesinde, gelecekteki gen tedavisinin faydalarından yararlanma olasılığı bulunmaktadır.