Kristalline Retinopathie ist ein Sammelbegriff für eine heterogene Gruppe von Erkrankungen mit Kristallablagerungen in einer beliebigen Schicht oder Region der Netzhaut. Die Ursachen sind vielfältig: Erbkrankheiten, Nebenwirkungen von Medikamenten, Komplikationen systemischer Erkrankungen usw.
Die wichtigsten erblichen kristallinen Retinopathien sind:
Bietti-Kristalline Netzhautdystrophie (BCD): autosomal-rezessive Erkrankung durch CYP4V2-Genmutation (4q35), 1937 von Bietti beschrieben. Die drei Hauptmerkmale sind verstreute glitzernde kristalline gelb-weiße Flecken im hinteren Pol, Atrophie der Choriokapillaris und kristalline Hornhautablagerungen, aber es wurden viele Fälle ohne Hornhautbeteiligung berichtet. Häufig in Ostasien, insbesondere bei Japanern und Chinesen. Etwa 10 % der Fälle, die als autosomal-rezessive nicht-syndromale RP diagnostiziert wurden, sind tatsächlich BCD.
Zystinose : Autosomal-rezessive lysosomale Speicherkrankheit durch CTNS-Genmutation (17p13.2).
Primäre Hyperoxalurie (PH) : Autosomal-rezessive Erkrankung durch AGXT/GRHPR/HOGA1-Genmutationen.
Sjögren-Larsson-Syndrom : Autosomal-rezessive Erkrankung durch ALDH3A2-Genmutation (17p11.2).
Die kristalline Retinopathie wird als verwandte Erkrankung der Retinitis pigmentosa eingestuft. 1)
QWas ist die Bietti-kristalline Netzhautdystrophie (BCD)?
A
Autosomal-rezessive Aderhaut-Netzhaut-Erkrankung durch CYP4V2-Genmutation, gekennzeichnet durch Kristallablagerungen im hinteren Pol, RPE- und Aderhautatrophie sowie Hornhautkristalle. Häufiger bei Ostasiaten, manifestiert sich mit Nachtblindheit und Gesichtsfeldausfällen im Alter von 20–40 Jahren und führt im Alter von 50–60 Jahren zu schwerer Sehbehinderung. Derzeit gibt es keine kausale Behandlung, Gentherapieforschung läuft.
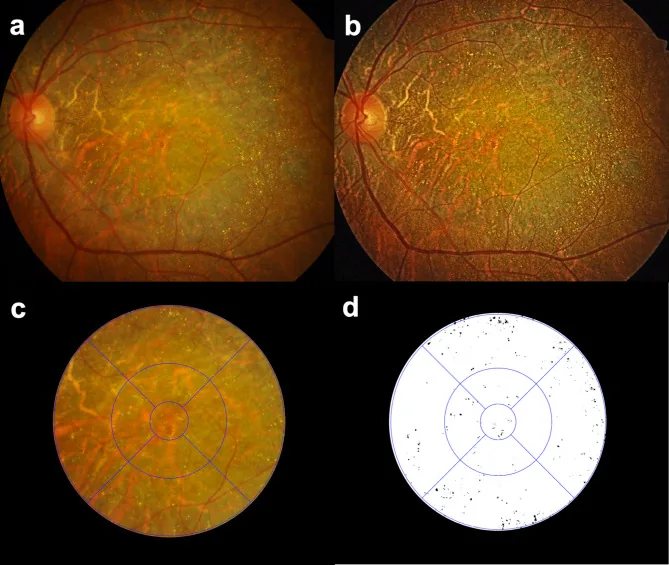
Seung Jun You; Chang Ki Yoon; Un Chul Park; Kyu Hyung Park; Eun Kyoung Lee. Longitudinal quantitative assessment of retinal crystalline deposits in bietti crystalline dystrophy. BMC Ophthalmol. 2025 Mar 17; 25:139. Figure 1. PMCID: PMC11916969. License: CC BY.
Quantitative Bewertung von kristallinen Netzhautablagerungen in Augen mit Bietti-kristalliner Dystrophie. (a) Farbfundusfoto mit grauweißer Aderhaut-Netzhaut-Atrophie und zahlreichen gelb-weißen Kristallablagerungen. (b) Bildkontrast verstärkt mittels kontrastbegrenzter adaptiver Histogramm-Egalisierung (CLAHE) zur besseren Erkennung von Netzhautkristallablagerungen. (c) ETDRS-Gitter überlagert zur regionalen Quantifizierung von Netzhautkristallablagerungen. (d) Netzhautkristalle extrahiert als Schwarz auf weißem Hintergrund mit Medilabel®-Software.
Die subjektiven Symptome der einzelnen Erkrankungen sind wie folgt. Sie zeigen progressive Gesichtsfeld- und Sehschärfenverluste, aber die Prognose variiert von Fall zu Fall. 1)
BCD :
Progressive Nachtblindheit und Gesichtsfeldausfälle (parazentrales Skotom) beginnen im Alter von 20–40 Jahren.
Danach rapider Sehschärfenverlust, der im Alter von 50–60 Jahren zu schwerer Sehbehinderung führt.
Die klinischen Befunde der BCD werden nach der 3-Stadien-Klassifikation von Yuzawa eingeteilt.
Die folgende Tabelle zeigt die Korrelation zwischen Kristallablagerungen und RPE-Atrophie in jedem Stadium.
Stadium
Kristallablagerungen
RPE-Atrophie
1
Zahlreich im hinteren Pol
Leicht, makulär
2
Vermindert im hinteren Pol
Progressiv, ausgedehnt
3
Fast verschwunden
Schwer, generalisiert
Stadium 1: Zahlreiche kleine, glänzende, gelb-weiße Kristalle verstreut vom hinteren Pol bis zur mittleren Peripherie. Sie befinden sich auf der Ebene des RPE-Choriokapillaris-Komplexes. Begleitende leichte makuläre RPE-Atrophie.
Stadium 2: Progressive RPE-Atrophie und Chorioretinalatrophie, die über den hinteren Pol hinausgeht. Die Kristalle nehmen im hinteren Pol ab und verbleiben in der mittleren Peripherie.
Stadium 3: Ausgedehnte RPE- und Choriokapillaris-Atrophie. Die Kristalle sind fast vollständig verschwunden.
Einige Fälle weisen Kristalle im vorderen Hornhautstroma nahe dem Limbus auf, aber sie sind nicht in allen Fällen vorhanden.
Iatrogen : Ablagerung infolge einer therapeutischen Maßnahme.
QWas sind die Ursachen einer kristallinen Retinopathie?
A
Die Hauptursachen sind erblich (BCD, Cystinose, Hyperoxalurie, Sjögren-Larsson-Syndrom) und medikamentös (Tamoxifen, Canthaxanthin, Talkum usw.). Behandlung und Prognose variieren stark je nach Ursache, daher ist eine genaue Ursachendiagnose für die Therapieplanung unerlässlich.
FA : In RPE-Atrophiebereichen zeigt sich eine Hyperfluoreszenz durch Fensterdefekte.
ICGA : Verzögerte Aderhautfüllung in allen Stadien. Spätphase zeigt plattenförmige Hypofluoreszenz.
OCT und FAF
SD-OCT : Hochreflektierende Punkte in allen Netzhautschichten (hauptsächlich im RPE-Bruch-Membran-Komplex). In der äußeren Körnerschicht können äußere Netzhauttubulusstrukturen (ORTs) auftreten.
FAF : Die Kristalle selbst sind unsichtbar. Geschädigte RPE-Zellen zeigen eine körnige starke Autofluoreszenz, während RPE-Atrophiebereiche eine geringe Autofluoreszenz aufweisen. Nahinfrarotlicht (NIR) stellt die Kristalle gut dar.
Elektroretinogramm
Funktionsbeurteilung : Je nach Stadium normal → vermindert → erloschen. Eine Zapfen-Stäbchen-Dysfunktion ist häufig.
Anwendung zur Differenzialdiagnose : Bei BCD fehlt eine Verengung der Netzhautgefäße, und die Elektroretinogramm-Antwort ist relativ erhalten, was zur Abgrenzung von Retinitis pigmentosa beiträgt. Gentests sind ebenfalls hilfreich.
Zystinose : Die Messung der freien, nicht proteingebundenen Zystinkonzentration in polymorphkernigen Leukozyten ist für die definitive Diagnose nützlich.
Primäre Hyperoxalurie : Bestätigung einer erhöhten Oxalatausscheidung (mindestens das Doppelte des Normalwerts) im 24-Stunden-Sammelurin. Gentests (AGXT/GRHPR/HOGA1) können die Diagnose ebenfalls bestätigen.
Sjögren-Larsson-Syndrom : Gentests (ALDH3A2) oder Messung der FALDH-Aktivität (fettaldehyddehydrogenase).
Tamoxifen-Retinopathie : Differenzialdiagnostisch sind CYP4V2-Kristallretinopathie, Drusen und parafoveale Teleangiektasien zu berücksichtigen.
QWie unterscheidet man eine kristalline Retinopathie von einer Retinitis pigmentosa?
A
Bei BCD fehlt eine Verengung der Netzhautgefäße, und die Elektroretinogramm-Antwort ist relativ erhalten. Gentests (CYP4V2) sind für die definitive Diagnose nützlich, und etwa 10 % der Fälle, die als autosomal-rezessive nicht-syndromale Retinitis pigmentosa diagnostiziert wurden, sind tatsächlich BCD.
Absetzen des Medikaments stoppt das Fortschreiten und führt manchmal zu einer Besserung.
Bei zystoidem Makulaödem können Anti-VEGF-Medikamente wirksam sein.
QSind die durch Tamoxifen verursachten kristallinen Ablagerungen in der Netzhaut reversibel?
A
Durch das Absetzen des Medikaments wird das Fortschreiten gestoppt und es kann gelegentlich zu einer Besserung kommen. Bei begleitendem zystoidem Makulaödem (CME) können Anti-VEGF-Medikamente wirksam sein. In beiden Fällen ist die Früherkennung der Schlüssel zur Verbesserung der Prognose.
6. Pathophysiologie und detaillierte Krankheitsmechanismen
BCD: CYP4V2 kodiert ein Enzym der Cytochrom-P450-Familie, das am Lipid- und Steroidstoffwechsel beteiligt ist. CYP4V2-Mutationen führen zur Ablagerung abnormaler Lipidmetaboliten in der Aderhaut und Netzhaut, was zur Atrophie des RPE und der Choriokapillaris sowie zur sekundären Degeneration der Photorezeptoren führt.
Zystinose: Die Funktionsstörung von Cystinosin (einem lysosomalen Membran-Cystin-Transporter) hemmt den Cystintransport aus den Lysosomen. Die Akkumulation von Cystin in retinalen Endothelzellen führt zur Netzhautdegeneration.
Primäre Hyperoxalurie: Eine angeborene Störung des Glyoxylatstoffwechsels in der Leber führt zu einer Überproduktion von Oxalat und Glykolat. Infolgedessen lagern sich Calciumoxalatkristalle in der Netzhaut, dem RPE und der Aderhaut ab, was zu einer fortschreitenden Netzhautdegeneration führt.
Sjögren-Larsson-Syndrom: Der Mangel an FALDH (fettaldehyddehydrogenase) führt zur Akkumulation von aliphatischen Aldehyden und Alkoholen, was die Müller-Zellen und Photorezeptoren schädigt.
Tamoxifen-Retinopathie: Tamoxifen bindet an Lipide und reichert sich in Lysosomen an, wodurch die Enzymaktivität verringert wird und kristalline Substanzen in der Nervenfaserschicht und der inneren plexiformen Schicht abgelagert werden.
Eine Phase-1-Studie zur Gentherapie für BCD mit rAAV2/8-hCYP4V2 (subretinale Injektion) ist im Gange. Frühe Gentests und genetische Beratung könnten den Zugang zu zukünftigen Gentherapien ermöglichen.