La retinopatia cristallina è un termine generico per un gruppo eterogeneo di malattie caratterizzate da depositi cristallini in qualsiasi strato o regione della retina. Le cause sono varie: malattie ereditarie, effetti collaterali di farmaci, complicanze di malattie sistemiche, ecc.
La classificazione delle cause è la seguente:
Ereditarie: distrofia cristallina retinica di Bietti (BCD), cistinosi, iperossaluria primaria, sindrome di Sjögren-Larsson
Le principali retinopatie cristalline ereditarie includono:
Distrofia cristallina retinica di Bietti (BCD): malattia autosomica recessiva dovuta a mutazioni del gene CYP4V2 (4q35), descritta da Bietti nel 1937. Le tre caratteristiche principali sono macchie giallo-biancastre cristalline scintillanti disseminate al polo posteriore, atrofia della coriocapillare e depositi cristallini corneali, ma sono stati riportati molti casi senza coinvolgimento corneale. Frequente in Asia orientale, specialmente tra giapponesi e cinesi. Circa il 10% dei casi diagnosticati come retinite pigmentosa non sindromica autosomica recessiva sono in realtà BCD.
Cistinosi : Malattia da accumulo lisosomiale autosomica recessiva dovuta a mutazioni del gene CTNS (17p13.2).
Iperossaluria primaria (PH) : Malattia autosomica recessiva dovuta a mutazioni dei geni AGXT/GRHPR/HOGA1.
Sindrome di Sjögren-Larsson : Malattia autosomica recessiva dovuta a mutazioni del gene ALDH3A2 (17p11.2).
La retinopatia cristallina è considerata una malattia correlata alla retinite pigmentosa. 1)
QCos'è la distrofia cristallina retinica di Bietti (BCD)?
A
Malattia corioretinica autosomica recessiva dovuta a mutazioni del gene CYP4V2, caratterizzata da depositi cristallini al polo posteriore, atrofia dell’EPR e della coroide e cristalli corneali. Più comune nelle popolazioni dell’Asia orientale, si manifesta con nictalopia e difetti del campo visivo tra i 20 e i 40 anni e porta a grave deficit visivo tra i 50 e i 60 anni. Attualmente non esiste una terapia curativa, ma sono in corso ricerche sulla terapia genica.
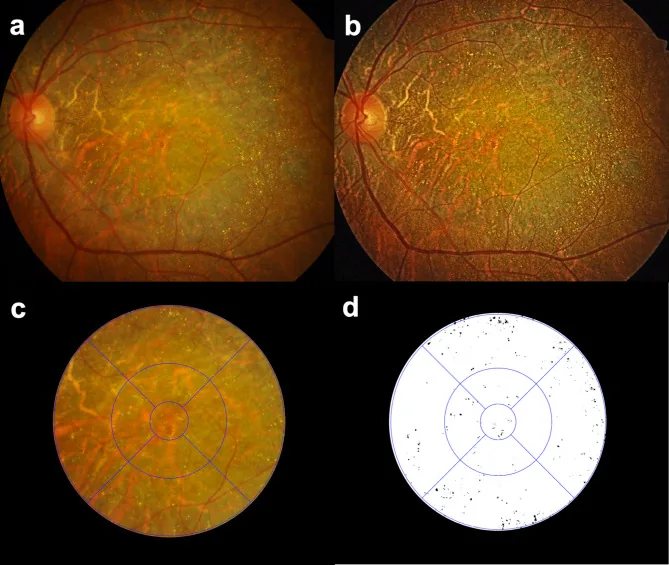
Seung Jun You; Chang Ki Yoon; Un Chul Park; Kyu Hyung Park; Eun Kyoung Lee. Longitudinal quantitative assessment of retinal crystalline deposits in bietti crystalline dystrophy. BMC Ophthalmol. 2025 Mar 17; 25:139. Figure 1. PMCID: PMC11916969. License: CC BY.
Valutazione quantitativa dei depositi cristallini retinici in occhi con distrofia cristallina di Bietti. (a) Fotografia a colori del fondo oculare che mostra atrofia corioretinica grigiastra e numerosi depositi cristallini giallo-bianchi. (b) Contrasto dell’immagine migliorato utilizzando l’equalizzazione dell’istogramma adattativa a contrasto limitato (CLAHE) per rilevare meglio i depositi cristallini retinici. (c) Griglia ETDRS sovrapposta per la quantificazione regionale dei depositi cristallini retinici. (d) Cristalli retinici estratti come nero su sfondo bianco utilizzando il software Medilabel®.
I sintomi soggettivi di ciascuna malattia sono i seguenti. Presentano progressivi deficit del campo visivo e dell’acuità visiva, ma la prognosi varia da caso a caso. 1)
BCD :
Nictalopia progressiva e difetti del campo visivo (scotoma paracentrale) che iniziano tra i 20 e i 40 anni.
Successivamente, rapido calo dell’acuità visiva, che porta a grave deficit visivo tra i 50 e i 60 anni.
Fotofobia e blefarospasmo dovuti a depositi di cistina nella cornea e nella congiuntiva nel primo anno di vita
Riduzione dell’acuità visiva e restringimento del campo visivo dovuti a degenerazione retinica progressiva
La cistinosi nefropatica infantile (95%) si presenta con ritardo della crescita e acidosi tubulare renale, portando a insufficienza renale nell’adolescenza
Iperossaluria primaria:
Calcoli renali ricorrenti e coliche renali compaiono nel 50% dei pazienti prima dei 5 anni
La riduzione dell’acuità visiva è più probabilmente dovuta ad atrofia ottica che a depositi cristallini retinici
Entro i 25 anni, il 90% dei pazienti presenta sintomi
I reperti clinici della BCD sono classificati secondo la classificazione a 3 stadi di Yuzawa.
La tabella seguente mostra la corrispondenza tra depositi cristallini e atrofia dell’EPR in ciascuno stadio.
Stadio
Depositi cristallini
Atrofia dell’EPR
1
Numerosi al polo posteriore
Lieve, maculare
2
Ridotti al polo posteriore
Progressiva, estesa
3
Quasi scomparsi
Grave, generalizzata
Stadio 1: Numerosi piccoli cristalli giallo-biancastri brillanti sparsi dal polo posteriore alla periferia media. Si trovano a livello del complesso EPR-coriocapillare. Associata lieve atrofia maculare dell’EPR.
Stadio 2: Atrofia progressiva dell’EPR e atrofia corioretinica che si estende oltre il polo posteriore. I cristalli diminuiscono al polo posteriore e persistono nella periferia media.
Stadio 3: Atrofia estesa dell’EPR e della coriocapillare. I cristalli sono quasi completamente scomparsi.
Alcuni casi presentano cristalli nello stroma corneale anteriore vicino al limbo, ma non sono presenti in tutti i casi.
Cristalli di ossalato gialli: distribuiti principalmente intorno alla fovea, nello strato plessiforme esterno e nello strato nucleare esterno intorno alle arterie
Lesioni anulari sottoretiniche nere dovute a proliferazione dell’RPE → progressione verso atrofia geografica
Viene anche valutata con il grading di Derveaux (gradi 1–4).
Grado 1 : cristalli di ossalato foveali isolati
Grado 2 : cristalli maculari con risparmio della fovea + proliferazione dell’RPE
Grado 3 : proliferazione dell’RPE, fibrosi sottoretinica e fibrosi foveale
BCD (CYP4V2) : autosomico recessivo. Frequente negli asiatici orientali. Anomalia degli enzimi del metabolismo lipidico e steroideo. 1)
Cistinosi (CTNS) : autosomico recessivo. 1 su 100.000-200.000 nati. Malattia da accumulo lisosomiale.
Iperossaluria (AGXT ecc.) : autosomico recessivo. Prevalenza inferiore a 3 persone per milione.
Sindrome di Sjögren-Larsson (ALDH3A2) : autosomico recessivo. Anomalia del metabolismo delle aldeidi alifatiche.
Farmaci
Tamoxifene : depositi cristallini nello strato delle fibre nervose e nello strato plessiforme interno.
Cantaxantina : depositi dovuti a colorante alimentare (integratore).
Talco : eccipiente di compresse. Può causare retinopatia da talco dopo somministrazione endovenosa.
Metossiflurano, nitrofurantoina : uso attualmente limitato.
Altri
Degenerazione : depositi cristallini associati a cambiamenti degenerativi cronici della retina.
Idiopatici : depositi cristallini di causa sconosciuta.
Iatrogeno : deposito conseguente a un atto terapeutico.
QQuali sono le cause della retinopatia cristallina?
A
Le cause principali sono ereditarie (BCD, cistinosi, iperossaluria, sindrome di Sjögren-Larsson) e farmacologiche (tamoxifene, cantaxantina, talco, ecc.). Trattamento e prognosi variano notevolmente a seconda della causa, pertanto una diagnosi eziologica accurata è essenziale per determinare la strategia terapeutica.
FA : Nelle aree di atrofia dell’EPR si osserva iperfluorescenza per difetto a finestra.
ICGA : Ritardo di riempimento coroideale in tutti gli stadi. Ipofluorescenza a placca nella fase tardiva.
OCT e FAF
SD-OCT : Punti iperriflettenti in tutti gli strati retinici (principalmente nel complesso RPE-membrana di Bruch). Nello strato nucleare esterno possono essere presenti strutture tubulari retiniche esterne (ORTs).
FAF : I cristalli stessi sono invisibili. Le cellule RPE danneggiate mostrano una autofluorescenza granulare intensa, mentre le aree di atrofia RPE mostrano ipoautofluorescenza. La luce nel vicino infrarosso (NIR) visualizza bene i cristalli.
Elettroretinogramma
Valutazione funzionale : Normale → ridotta → assente a seconda dello stadio. La disfunzione di tipo cono-bastoncello è comune.
Applicazione alla diagnosi differenziale : Nella BCD non c’è restringimento dei vasi retinici e la risposta elettroretinografica è relativamente preservata, il che aiuta a differenziarla dalla retinite pigmentosa. Anche i test genetici sono utili.
Cistinosi : La misurazione della concentrazione di cistina libera non legata alle proteine nei leucociti polimorfonucleati è utile per la diagnosi definitiva.
Iperossaluria primaria : Conferma di un aumento dell’escrezione di ossalato (almeno il doppio del normale) nelle urine delle 24 ore. I test genetici (AGXT/GRHPR/HOGA1) possono anche confermare la diagnosi.
Sindrome di Sjögren-Larsson : Test genetici (ALDH3A2) o misurazione dell’attività FALDH (aldeide deidrogenasi alifatica).
Retinopatia da tamoxifene : La diagnosi differenziale include la retinopatia cristallina CYP4V2, le drusen e la teleangectasia parafoveale.
QCome si distingue la retinopatia cristallina dalla retinite pigmentosa?
A
Nella BCD non c’è restringimento dei vasi retinici e la risposta elettroretinografica è relativamente preservata. I test genetici (CYP4V2) sono utili per la diagnosi definitiva e circa il 10% dei casi diagnosticati come retinite pigmentosa non sindromica autosomica recessiva sono in realtà BCD.
Attualmente non esiste una terapia curativa efficace. La cura dell’ipovisione è fondamentale e per le complicanze viene effettuata la seguente gestione:
QI depositi cristallini retinici da tamoxifene sono reversibili?
A
La sospensione del farmaco arresta la progressione e talvolta porta a un miglioramento. In caso di edema maculare cistoide (CME) associato, i farmaci anti-VEGF possono essere efficaci. In entrambi i casi, la diagnosi precoce è la chiave per migliorare la prognosi.
I meccanismi di ciascuna malattia sono i seguenti:
BCD: CYP4V2 codifica un enzima della famiglia del citocromo P450, coinvolto nel metabolismo dei lipidi e degli steroidi. Le mutazioni di CYP4V2 portano al deposito di metaboliti lipidici anomali nella coroide e nella retina, causando atrofia dell’EPR e della coriocapillare, e successiva degenerazione secondaria dei fotorecettori.
Cistinosi: La disfunzione della cistinosina (proteina di trasporto della cistina nella membrana lisosomiale) inibisce il trasporto della cistina fuori dai lisosomi. L’accumulo di cistina nelle cellule endoteliali retiniche causa degenerazione retinica.
Iperossaluria primaria: Un’anomalia congenita del metabolismo del gliossilato nel fegato porta a un’eccessiva produzione di ossalato e glicolato. Di conseguenza, cristalli di ossalato di calcio si depositano nella retina, nell’EPR e nella coroide, causando una degenerazione retinica progressiva.
Sindrome di Sjögren-Larsson: Il deficit di FALDH (aldeide deidrogenasi alifatica) porta all’accumulo di aldeidi e alcoli alifatici, danneggiando le cellule di Müller e i fotorecettori.
Retinopatia da tamoxifene: Il tamoxifene si lega ai lipidi e si accumula nei lisosomi, riducendo l’attività enzimatica e causando il deposito di sostanze cristalline nello strato delle fibre nervose e nello strato plessiforme interno.
È in corso uno studio clinico di fase 1 per la terapia genica sostitutiva per BCD con rAAV2/8-hCYP4V2 (iniezione sottoretinica). Un test genetico precoce e una consulenza genetica potrebbero consentire di beneficiare di future terapie geniche.