Crystalline retinopathy is a general term for a heterogeneous group of diseases characterized by crystal deposits in any layer or region of the retina. Causes are diverse, including hereditary diseases, drug side effects, and complications of systemic diseases.
Representative hereditary crystalline retinopathies include the following:
Bietti crystalline retinal dystrophy (BCD): An autosomal recessive disorder caused by mutations in the CYP4V2 gene (4q35), first reported by Bietti in 1937. The three main features are scattered glittering crystal-like yellowish-white spots in the posterior pole, atrophy of the choriocapillaris, and crystal-like deposits in the cornea, although many cases without corneal involvement have been reported. It is more common in East Asia, especially in Japanese and Chinese populations. It accounts for 10% of cases diagnosed as autosomal recessive non-syndromic retinitis pigmentosa.
Cystinosis: An autosomal recessive lysosomal storage disease caused by mutations in the CTNS gene (17p13.2).
Primary hyperoxaluria (PH): An autosomal recessive disorder caused by mutations in the AGXT/GRHPR/HOGA1 genes.
Sjögren-Larsson syndrome: An autosomal recessive disorder caused by mutations in the ALDH3A2 gene (17p11.2).
Crystalline retinopathy is considered a related disease to retinitis pigmentosa. 1)
QWhat is Bietti crystalline dystrophy (BCD)?
A
It is an autosomal recessive chorioretinal disease caused by mutations in the CYP4V2 gene, characterized by crystalline deposits in the posterior pole, RPE and choroidal atrophy, and corneal crystals. It is more common in East Asian populations, presenting with night blindness and visual field defects in the 20s to 40s, leading to severe visual impairment in the 50s to 60s. Currently, there is no curative treatment, but gene therapy research is ongoing.
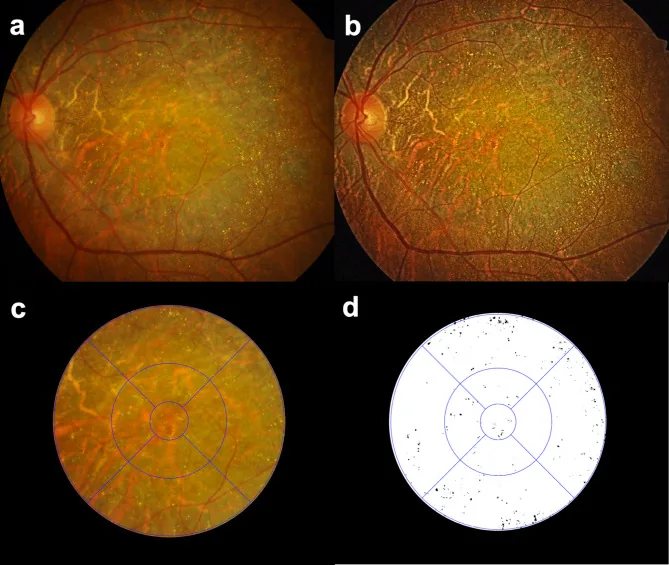
Seung Jun You; Chang Ki Yoon; Un Chul Park; Kyu Hyung Park; Eun Kyoung Lee. Longitudinal quantitative assessment of retinal crystalline deposits in bietti crystalline dystrophy. BMC Ophthalmol. 2025 Mar 17; 25:139. Figure 1. PMCID: PMC11916969. License: CC BY.
Quantitative assessment of retinal crystalline deposits in eyes with Bietti Crystalline Dystrophy. (a) A color fundus photograph showing greyish chorioretinal atrophy and numerous yellow-white crystalline deposits. (b) Image contrast enhanced using Contrast Limited Adaptive Histogram Equalization (CLAHE) to better detect retinal crystalline deposits. (c) An Early Treatment of Diabetic Retinopathy Study (ETDRS) grid overlaid for regional quantification of retinal crystalline deposits. (d) Retinal crystals extracted as black on a white background using Medilabel® software
The subjective symptoms of each disease are as follows. They present with progressive visual field and visual acuity loss, but the prognosis varies among cases. 1)
BCD:
Progressive night blindness and visual field defects (paracentral scotomas) develop in the 20s to 40s.
Subsequently, rapid visual acuity loss occurs, leading to severe visual impairment in the 50s to 60s.
The clinical findings of BCD are organized according to Yuzawa’s three-stage classification.
The correspondence of crystal deposition and RPE atrophy in each stage is shown below.
Stage
Crystal deposits
RPE atrophy
1
Numerous in posterior pole
Mild, macular
2
Decreased in posterior pole
Progressive, extensive
3
Almost absent
Severe, diffuse
Stage 1: Numerous small, shiny yellowish-white crystals scattered from the posterior pole to the mid-periphery. They are located at the level of the RPE-choriocapillaris complex. Associated with mild macular RPE atrophy.
Stage 2: Progressive RPE atrophy and chorioretinal atrophy extending beyond the posterior pole. Crystals decrease in the posterior pole and remain in the mid-periphery.
Stage 3: Extensive RPE and choriocapillaris atrophy. Crystals are almost absent.
Some cases show corneal stromal crystals in the anterior peripheral cornea, but they are not present in all cases.
Tamoxifen: Crystalline deposits in the nerve fiber layer and inner plexiform layer.
Canthaxanthin: Deposits from food coloring (supplements).
Talc: Tablet excipient. Can cause talc retinopathy after intravenous administration.
Methoxyflurane and nitrofurantoin: Currently limited use.
Other
Degeneration: Crystalline deposits associated with chronic degenerative changes in the retina.
Idiopathic: Crystal deposits of unknown cause.
Iatrogenic: Deposition resulting from medical treatment.
QWhat are the causes of crystalline retinopathy?
A
The main causes are hereditary (BCD, cystinosis, hyperoxaluria, Sjögren-Larsson syndrome) and drug-induced (tamoxifen, canthaxanthin, talc, etc.). Since treatment and prognosis vary greatly depending on the cause, accurate diagnosis of the cause is essential for determining the treatment strategy.
FA: Hyperfluorescence due to window defects is observed in areas of RPE atrophy.
ICGA: Delayed choroidal filling in all stages. Late-phase shows patchy hypofluorescence.
OCT and FAF
SD-OCT: Hyperreflective dots in all retinal layers (mostly in the RPE-Bruch membrane complex). Outer retinal tubulations (ORTs) may be seen in the outer nuclear layer.
FAF: Crystals themselves are not visible. Damaged RPE cells show granular hyperautofluorescence, while areas of RPE atrophy show hypoautofluorescence. Near-infrared (NIR) imaging visualizes crystals well.
Electroretinography
Functional assessment: Normal → decreased → absent depending on stage. Cone-rod pattern dysfunction is common.
Application in differential diagnosis: In BCD, there is no narrowing of retinal vessels, and the electroretinogram response is relatively preserved, which helps differentiate it from retinitis pigmentosa. Genetic testing is also useful.
Cystinosis: Measurement of free non-protein-bound cystine concentration in polymorphonuclear leukocytes is useful for definitive diagnosis.
Primary hyperoxaluria: Increased oxalate excretion (more than twice normal) in 24-hour urine collection. Genetic testing (AGXT/GRHPR/HOGA1) can also confirm.
Sjögren-Larsson syndrome: Genetic testing (ALDH3A2) or measurement of FALDH (fatty aldehyde dehydrogenase) activity.
Tamoxifen retinopathy: Consider CYP4V2 crystalline retinopathy, drusen, and parafoveal telangiectasia in the differential diagnosis.
QHow is crystalline retinopathy distinguished from retinitis pigmentosa?
A
In BCD, there is no narrowing of retinal vessels, and the electroretinogram response is relatively preserved. Genetic testing (CYP4V2) is useful for definitive diagnosis, and approximately 10% of cases diagnosed as autosomal recessive non-syndromic retinitis pigmentosa are actually BCD.
QCan retinal crystal deposits caused by tamoxifen be cured?
A
Discontinuation of the drug stops progression and sometimes leads to improvement. If cystoid macular edema (CME) is present, anti-VEGF agents may be effective. In all cases, early detection is key to improving prognosis.
BCD: CYP4V2 encodes a cytochrome P450 family enzyme involved in lipid and steroid metabolism. CYP4V2 mutations cause abnormal lipid metabolites to deposit in the chorioretina, leading to atrophy of the RPE and choriocapillaris, and secondary photoreceptor degeneration.
Cystinosis: Dysfunction of cystinosin (lysosomal membrane cystine transporter) impairs cystine transport out of lysosomes. Cystine accumulation in retinal endothelial cells causes retinal degeneration.
Primary hyperoxaluria: Congenital abnormalities in hepatic glyoxylate metabolism lead to overproduction of oxalate and glycolate. Consequently, calcium oxalate crystals deposit in the retina, RPE, and choroid, leading to progressive retinal degeneration.
Sjögren-Larsson syndrome: Deficiency of FALDH (fatty aldehyde dehydrogenase) leads to accumulation of fatty aldehydes and alcohols, causing damage to Müller cells and photoreceptors.
Tamoxifen retinopathy: Tamoxifen binds to lipids and accumulates in lysosomes, reducing enzyme activity and causing crystalline deposits in the nerve fiber layer and inner plexiform layer.
A phase 1 clinical trial of rAAV2/8-hCYP4V2 (subretinal administration) as gene replacement therapy for BCD is ongoing. Early genetic testing and genetic counseling may allow patients to benefit from future gene therapy.