Retinopatia cristalina (Crystalline retinopathy) é um termo abrangente para um grupo heterogêneo de doenças caracterizadas por depósitos cristalinos em qualquer camada ou região da retina. As causas são variadas, incluindo doenças genéticas, efeitos colaterais de medicamentos e complicações de doenças sistêmicas.
A classificação das causas é a seguinte:
Genéticas: Distrofia retiniana cristalina de Bietti (BCD), cistinose, hiperoxalúria primária, síndrome de Sjögren-Larsson
Exemplos representativos de retinopatia cristalina genética:
Distrofia retiniana cristalina de Bietti (BCD): Doença autossômica recessiva causada por mutação no gene CYP4V2 (4q35). Relatada pela primeira vez por Bietti em 1937. As três principais características são manchas amarelo-esbranquiçadas cintilantes cristalinas dispersas no polo posterior, atrofia da lâmina coriocapilar e depósitos cristalinos na córnea, mas muitos casos sem envolvimento corneano foram relatados. Comum no Leste Asiático, especialmente japoneses e chineses. Estima-se que 10% dos casos de RP não sindrômica autossômica recessiva sejam, na verdade, esta doença.
Cistinose: Doença de armazenamento lisossômico autossômica recessiva devido à mutação no gene CTNS (17p13.2).
Hiperoxalúria Primária (PH): Autossômica recessiva devido a mutações nos genes AGXT/GRHPR/HOGA1.
Síndrome de Sjögren-Larsson: Autossômica recessiva devido à mutação no gene ALDH3A2 (17p11.2).
QO que é a Distrofia Retiniana Cristalina de Bietti (BCD)?
A
É uma doença coriorretiniana autossômica recessiva devido à mutação no gene CYP4V2, caracterizada por depósitos cristalinos no polo posterior, atrofia do EPR e coroide, e cristais na córnea. É mais comum em asiáticos orientais, manifestando-se entre 20-40 anos como cegueira noturna e defeitos de campo visual, levando a deficiência visual grave entre 50-60 anos. Atualmente não há tratamento curativo; pesquisas de terapia gênica estão em andamento.
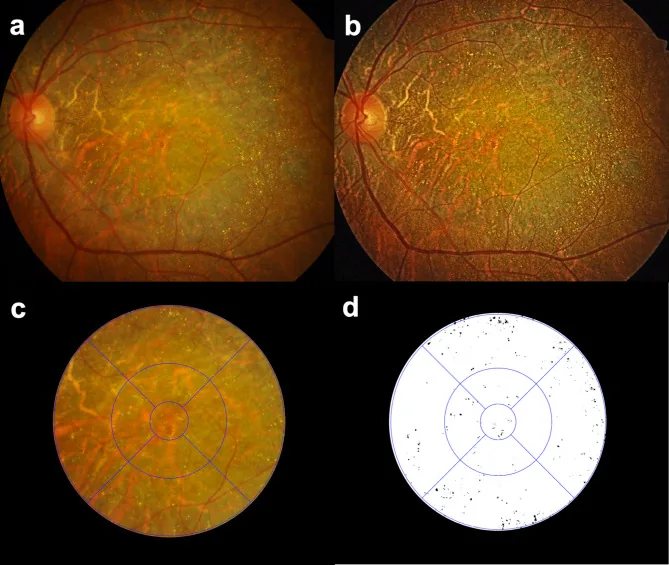
Seung Jun You; Chang Ki Yoon; Un Chul Park; Kyu Hyung Park; Eun Kyoung Lee. Longitudinal quantitative assessment of retinal crystalline deposits in bietti crystalline dystrophy. BMC Ophthalmol. 2025 Mar 17; 25:139. Figure 1. PMCID: PMC11916969. License: CC BY.
Avaliação quantitativa dos depósitos cristalinos retinianos em olhos com Distrofia Cristalina de Bietti. (a) Fotografia colorida do fundo de olho mostrando atrofia coriorretiniana acinzentada e numerosos depósitos cristalinos amarelo-esbranquiçados. (b) Contraste da imagem aprimorado usando Equalização de Histograma Adaptativa Limitada por Contraste (CLAHE) para melhor detectar depósitos cristalinos retinianos. (c) Grade do Estudo de Tratamento Precoce da Retinopatia Diabética (ETDRS) sobreposta para quantificação regional dos depósitos cristalinos retinianos. (d) Cristais retinianos extraídos como preto em fundo branco usando o software Medilabel®
Os sintomas subjetivos de cada doença são os seguintes. Ocorrem defeitos progressivos de campo visual e acuidade visual, mas o prognóstico varia conforme o caso. 1)
BCD:
Cegueira noturna progressiva e defeitos de campo visual (escotoma paracentral) surgem entre 20-40 anos
Em seguida, declínio acentuado da acuidade visual, levando a deficiência visual grave entre 50-60 anos
A rápida diminuição da acuidade visual pode ser devida a edema macular cistóide (EMC), neovascularização de coroide (NVC) ou buraco macular
No primeiro ano de vida, fotofobia e blefaroespasmo devido à deposição de cistina na córnea e conjuntiva
Diminuição da acuidade visual e estreitamento do campo visual devido à degeneração retiniana progressiva
Na cistinose nefropática infantil (95%), ocorre retardo do crescimento e acidose tubular renal, levando à insuficiência renal na segunda década de vida
Hiperoxalúria primária:
Cálculos renais recorrentes e cólica renal aparecem em 50% antes dos 5 anos de idade
A diminuição da acuidade visual é mais provavelmente causada por atrofia do nervo óptico do que por deposição de cristais na retina
Os achados clínicos da BCD são organizados pela classificação de 3 estágios de Yuzawa.
Abaixo está a correspondência da deposição de cristais e atrofia do EPR em cada estágio.
Estágio
Depósitos cristalinos
Atrofia do EPR
1
Numerosos no polo posterior
Leve na mácula
2
Diminuídos no polo posterior
Progressiva e extensa
3
Quase ausentes
Grave e generalizada
Estágio 1: Numerosos cristais amarelo-esbranquiçados pequenos e brilhantes dispersos do polo posterior até a periferia média. Localizados no nível do complexo EPR-coriocapilar. Acompanhados de atrofia leve do EPR macular.
Estágio 2: Atrofia progressiva do EPR e atrofia coriorretiniana além do polo posterior. Os cristais diminuem no polo posterior e permanecem na periferia média.
Estágio 3: Atrofia extensa do EPR e da coriocapilar. Os cristais quase desaparecem.
Alguns casos apresentam cristais no estroma corneano anterior próximo ao limbo, mas nem todos os casos os apresentam.
Cristais de oxalato amarelos: distribuídos principalmente ao redor da fóvea, na camada plexiforme externa e camada nuclear externa ao redor das artérias
Lesão anelar preta sub-retiniana devido à proliferação do EPR → progride para atrofia geográfica
Também é avaliada pela classificação de Derveaux (graus 1 a 4).
Grau 1: Cristais de oxalato isolados ao redor da fóvea
Grau 2: Cristais maculares poupando a fóvea + proliferação do EPR
Grau 3: Proliferação do EPR, fibrose sub-retiniana, fibrose foveal
BCD (CYP4V2): Autossômico recessivo. Comum em asiáticos orientais. Anomalia de enzimas do metabolismo lipídico/esteroide. 1)
Cistinose (CTNS): Autossômico recessivo. 1 em 100.000–200.000 nascimentos. Doença de armazenamento lisossômico.
Hiperoxalúria (AGXT etc.): Autossômico recessivo. Prevalência <3 por 1.000.000 de pessoas.
Síndrome de Sjögren-Larsson (ALDH3A2): Autossômico recessivo. Anomalia do metabolismo de aldeídos alifáticos.
Medicamentoso
Tamoxifeno: Depósito cristalino na camada de fibras nervosas e camada plexiforme interna.
Cantaxantina: Depósito devido a corante alimentar (suplemento).
Talco: Excipiente de comprimidos. Pode causar retinopatia por talco após administração intravenosa.
Metoxiflurano e Nitrofurantoína: Atualmente de uso limitado.
Outros
Degenerativo: Depósito cristalino associado a alterações degenerativas crônicas da retina.
Idiopático: Depósito cristalino de causa desconhecida.
Iatrogênico: Deposição resultante de procedimentos terapêuticos.
QQuais são as causas da retinopatia cristalina?
A
As principais causas são genéticas (BCD, cistinose, hiperoxalúria, síndrome de Sjögren-Larsson) e medicamentosas (tamoxifeno, cantaxantina, talco, etc.). O tratamento e o prognóstico variam muito conforme a causa, portanto, o diagnóstico preciso da causa é essencial para determinar o plano de tratamento.
Angiografia Fluoresceínica e Angiografia com Indocianina Verde
Angiografia Fluoresceínica: Mostra hiperfluorescência devido a defeitos em janela em áreas de atrofia do EPR.
Angiografia com Indocianina Verde: Atraso no preenchimento coroidal em todos os estágios. Mostra hipofluorescência em placa na fase tardia.
OCT e FAF
SD-OCT: Pontos hiperrefletivos em todas as camadas da retina (a maioria no complexo EPR-membrana de Bruch). Podem ser observadas estruturas tubulares retinianas externas (ORTs) na camada nuclear externa.
FAF: Os cristais em si não são visíveis. Células EPR danificadas mostram autofluorescência granular forte, enquanto áreas de atrofia do EPR mostram baixa autofluorescência. A luz infravermelha próxima (NIR) delineia bem os cristais.
Eletrorretinografia
Avaliação funcional: Normal → diminuída → ausente conforme o estágio. Disfunção do padrão cone-bastonete é comum.
Aplicação no diagnóstico diferencial: Na BCD, não há estreitamento dos vasos retinianos e a resposta eletrorretinográfica está relativamente preservada, o que ajuda a diferenciar da retinose pigmentar. O teste genético também é útil.
Cistinose: A medição da concentração de cistina livre não ligada a proteínas em leucócitos polimorfonucleares é útil para o diagnóstico definitivo.
Hiperoxalúria primária: Confirmação do aumento da excreção de oxalato na urina de 24 horas (mais que o dobro do normal). O teste genético (AGXT/GRHPR/HOGA1) também pode confirmar.
Síndrome de Sjögren-Larsson: Teste genético (ALDH3A2) ou medição da atividade da FALDH (aldeído graxo desidrogenase).
Retinopatia por tamoxifeno: No diagnóstico diferencial, considerar retinopatia cristalina por CYP4V2, drusas e telangiectasia parafoveal.
QComo diferenciar retinopatia cristalina de retinose pigmentar?
A
Na BCD, não há estreitamento dos vasos retinianos e a resposta eletrorretinográfica está relativamente preservada. O teste genético (CYP4V2) é útil para o diagnóstico definitivo, e estima-se que cerca de 10% dos casos diagnosticados como retinose pigmentar não sindrômica autossômica recessiva são, na verdade, BCD.
Atualmente, não existe terapia curativa eficaz. O cuidado básico é a reabilitação visual para baixa visão, e as complicações são gerenciadas da seguinte forma.
A interrupção do medicamento interrompe a progressão e, às vezes, melhora.
No edema macular cistóide, os medicamentos anti-VEGF podem ser eficazes.
QOs depósitos cristalinos na retina causados pelo tamoxifeno desaparecem?
A
Com a interrupção da administração, a progressão cessa e às vezes há melhora. Se houver edema macular cistóide (CME), os medicamentos anti-VEGF podem ser eficazes. Em ambos os casos, a detecção precoce é a chave para melhorar o prognóstico.
BCD: CYP4V2 codifica uma enzima da família do citocromo P450 envolvida no metabolismo de lipídios e esteroides. Mutações no CYP4V2 levam ao acúmulo de produtos anormais do metabolismo lipídico na coroide e retina, causando atrofia do EPR e da lâmina capilar coroidal, e degeneração secundária dos fotorreceptores.
Cistinose: A disfunção da cistinosina (proteína transportadora de cistina na membrana lisossomal) inibe o transporte de cistina para fora do lisossomo. O acúmulo de cistina nas células do sistema reticuloendotelial causa degeneração retiniana.
Hiperoxalúria Primária: Uma anomalia congênita do metabolismo do glioxilato no fígado leva à produção excessiva de oxalato e glicolato. Como resultado, cristais de oxalato de cálcio depositam-se na retina, EPR e coroide, progredindo a degeneração retiniana.
Síndrome de Sjögren-Larsson: A deficiência de FALDH (aldeído desidrogenase alifática) causa acúmulo de aldeídos e álcoois alifáticos, resultando em danos às células de Müller e fotorreceptores.
Retinopatia por Tamoxifeno: O tamoxifeno liga-se a lipídios e acumula-se nos lisossomos, reduzindo a atividade enzimática, levando à deposição de substâncias cristalinas na camada de fibras nervosas e na camada plexiforme interna.
Como terapia de reposição genética para BCD, o ensaio clínico de fase 1 do rAAV2/8-hCYP4V2 (injeção sub-retiniana) está em andamento. Com testes genéticos precoces e aconselhamento genético, há potencial para se beneficiar da terapia genética no futuro.