杜安氏症候群(DRS )是一種非進行性斜視 症候群,其本質為外展神經(CN6)先天性缺損 ,以及動眼神經對外直肌的異常支配。
這是最常見的先天性腦神經異常支配疾病(CCDD ),發生率約為一般人口的千分之一2) 。
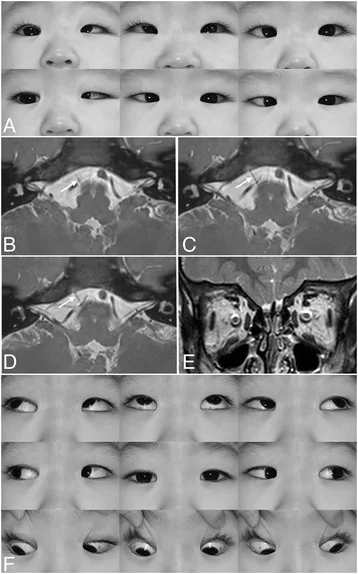
內轉時的眼球後退和眼瞼裂狹小是極具特徵的表現。
第一型(外轉限制為主)最常見,約佔整體的85%。多數在10歲前被診斷。
大多數患者雙眼視功能正常,並透過代償性頭位(臉部旋轉)維持雙眼視,視力 障礙較少。
手術並非眼球運動障礙 的根本治療,而是以矯正第一眼位偏位及改善頭位為目的。
杜安氏症候群(Duane Syndrome; Duane Retraction Syndrome, DRS )是1905年由Alexander Duane詳細描述的先天性、非進行性斜視 症候群。也稱為Stilling-Türk-Duane症候群 ,在ICD-10中歸類為H50.81。這是一種先天性眼球運動障礙 ,特徵為嚴重的內轉障礙,以及內轉時的眼球後退和由此引起的眼瞼裂狹小。
一般人口中約每1,000人中有1人發生2) ,佔所有斜視 病例的4%。是先天性腦神經異常支配疾病(CCDD )中最常見的疾病2) 。單側性佔82%,以左眼較多(59%),男女比約4:6,女性略多。雙側性約佔15-20%。多數在10歲前被診斷。
I型(約85%)
特徵 :外展受限 > 內收受限。正視時可能呈現內斜視 。
頭位 :最常見為向患側轉臉。72%有轉臉(68%轉向患側)1)
MRI所見 :約80%患者缺乏外展神經。
流行病學 :Anvari(125例)中I型佔87.0%1)
II型與III型
II型(5〜10%) :內收受限 > 外展受限。正視時呈現外斜視 ,轉臉方向與I型相反。MRI顯示大多數病例存在外展神經。
III型(10〜20%) :內收和外展均受限。93%有轉臉(73%轉向患側對側)1)
大規模研究(441名)中,54.6%出現臉部旋轉,單側性臉部旋轉的發生率顯著較高1) 。水平偏位76.0%,內斜視 58.4%,外斜視 17.6%1) 。
Q
杜安氏症候群有多常見?
A
一般人口中約每1,000人中有1人發生2) ,佔所有斜視 的4%。在先天性腦神經異常支配疾病(CCDD )中最常見。單側性佔82%,左眼和女童略多。多數在10歲前診斷。
Kim JH, et al. Postoperative full abduction in a patient of Duane retraction syndrome without an abducens nerve: a case report. BMC Ophthalmol. 2017. Figure 1. PMCID: PMC5438545. License: CC BY.
左側杜安氏症候群的多方向視圖,可見左眼外展受限、內收時眼裂狹窄及輕微上衝。對應本文「主要症狀與臨床發現」中討論的特徵性眼球運動異常。
雙眼視功能 :大多數病例雙眼視功能正常,很少造成視力 障礙。代償性頭位旋轉 :針對外展障礙,將臉轉向患側以維持雙眼視覺。必須向家長和教師充分說明,這種頭位異常是為了代償眼球運動障礙 、實現雙眼視覺所必需的行為。弱視 弱視 發生率為5.1%1) 。屈光 異常遠視 。據報告遠視 佔42.3%,近視 佔39.7%1) 。由於常合併屈光 異常,可能需要治療不等視弱視 。
外展受限 :完全或部分外展受限(程度因類型而異)。眼球後退+眼裂狹窄 :內轉時眼球後退,眼裂變窄(誘發性眼瞼下垂 )。這是DRS 的特徵性表現,從側面觀察容易確認。若內轉時眼裂狹窄不明顯,可從側面觀察眼球並使其內轉,更容易看出眼球後退。眼裂擴大 :外轉時眼裂擴大。上射/下射(牽制現象) :內轉時眼球向上或向下偏移。出生時不會出現,而是由於外直肌進行性攣縮繼發產生。內轉時,若外直肌向上偏移則產生上射,向下偏移則產生下射。輻輳不全
30%至50%的患者存在因先天性腦幹障礙引起的異常。具體例子包括鱷魚淚(味覺性流淚)和感音神經性聽力損失 。
相關症候群包括Duane radial ray症候群(SALL4突變)、Goldenhar症候群、HOXA1症候群等2) 。
Q
代償性面部旋轉需要治療嗎?
A
代償性頭位旋轉是維持雙眼視覺功能的適應性動作。若小於15度,原則上不需治療,僅需觀察。若大於15度或影響日常生活及美觀,則考慮手術。在學校,可安排兒童坐在面向旋轉方向的位置,進行環境調整。
外展神經(CN6)缺損 :胚胎發育4至8週時受損,導致外展神經運動神經元缺損 或發育不全。患側腦幹的外展神經核及外展神經發育不全或缺失。動眼神經(CN3)異常支配 :CN3內轉支異常支配外直肌,導致內轉時內直肌與外直肌同時收縮,造成眼球後退。眼瞼裂狹窄是由於眼球後退被動產生。外直肌的變化 :組織學上,外直肌的一部分(未接受正常神經支配的部分)發生纖維化。CCDD 之一2) 。
單獨型中10%為遺傳性,90%為散發性。I型為體染色體顯性(8q13),II型為體染色體顯性(DURS2: CHN1突變,2q31-q32.1)。主要基因突變如下2) 。
CHN1突變 :α2-chimaerin(Rac-GAP蛋白)的錯義突變。破壞蛋白質閉合結構的維持殘基,導致RacGAP活性過度亢進。CHN1突變病例多為雙側DRS ,可能伴隨垂直方向異常。MAFB突變 :雜合性功能喪失突變導致散發性DRS 。部分病例表現為DRS 合併聽力喪失及局部節段性腎小球硬化症(FSGS)。SALL4突變 :Duane radial ray症候群(DRS 合併上肢畸形),體染色體顯性。HOXA1突變 :雙側DRS 合併感覺神經性聽力損失、顏面神經麻痺、中樞性換氣不足、血管畸形及智能障礙。COL25A1突變 :先天性眼瞼下垂 或DRS (同一家族內表現型可能不同)。
遵循標準斜視 評估(視力 、眼球運動、雙眼視覺、屈光 檢查)。
裂隙燈 與眼球運動檢查 DRS 特徵。從側面觀察較易確認眼球後退。眼球牽引試驗 :陽性(中樞性但外眼肌有攣縮變化)。有助於鑑別機械性限制與神經支配缺損 。MRI :可顯示外展神經缺損 或發育不全。第一型80%患者MRI顯示外展神經缺損 ,而第二型多數患者外展神經存在。常規診斷不建議,但對術前評估有幫助。基因檢測(CHN1) :僅建議用於家族性病例。
疾病 鑑別要點 先天性外展神經麻痺 無眼球後退,內斜視 角較大 HGPPS 水平注視麻痺+進行性脊柱側彎,無眼球後退2) Moebius症候群 CN6+CN7障礙,伴隨臉部肌肉無力2) Brown症候群 以上轉受限為主,無眼球後縮
許多病例無需治療。眼球運動障礙 本身無根本治療方法。
屈光 矯正(眼鏡)遠視 即可消除臉部旋轉及內斜視 的案例存在1) 。優先治療屈光 異常或不等視弱視 。稜鏡眼鏡 弱視 治療弱視 ,進行標準遮蓋療法 。許多Duane症候群 患者藉由轉臉來維持雙眼視覺,因此斜視 性弱視 的風險不高。肉毒桿菌毒素注射 1) 。Ameri 的 16 名患者中,面部旋轉 18.27°±7.29° 在一週後改善至 0.094°,但六個月後又回升至 7°1) 。
手術適應症 :異常頭位大於 15°、第一眼位明顯偏位、重度誘發性眼瞼下垂 (眼瞼裂寬度減少 50% 以上)。
內斜視 DRS
內直肌後徙術 為基本術式。對於 I 型 Duane 症候群且內斜視 或異常頭位嚴重者有效。
內斜視 小於 15Δ 時,可採用單眼內直肌後徙 6mm 處理。若超過此度數,則需進行雙眼內直肌後徙。
注意後徙超過 5mm 可能導致內轉限制及續發性外斜視 的風險。
Pressman 的 19 名患者(術前 26.28PD → 術後 2.71PD,成功率 79%)1)
有報告指出,西田法合併內直肌後徙可將40PD的內斜視 改善至6PD,外展恢復至45°1) 。
重症・難治病例
對於上轉/下轉(upshoot/downshoot) ,可施行外直肌後徙或外直肌Y字分割術(Y-Splitting)。無論如何都無法完全治癒,因此需謹慎決定適應症。
外直肌鞏膜固定 (或大量後徙)合併內直肌後徙亦有效。
垂直直肌轉位術 (VRT) :將上、下直肌移至外直肌附著處。加強縫合組AHP從22.7°改善至3.6°,非加強組從18.7°改善至7°1) 。
手術並非DRS 的根本治療。神經支配缺陷在術後仍持續存在。
術前應說明手術目的為矯正第一眼位偏位、改善頭位、減輕誘發性眼瞼下垂 。
內直肌過度後徙有內轉限制及續發性外斜視 的風險。
Q
肉毒桿菌毒素注射的效果如何?
A
肉毒桿菌毒素可暫時改善異常頭位及內斜視 ,但效果在6個月後減弱1) 。作為手術的替代或輔助療法,可考慮用於嬰幼兒早期介入或手術風險較高的病例。有時需要重複注射。
Q
手術能根治杜安氏症候群嗎?
A
外展神經先天性缺損 及動眼神經異常支配的根本神經障礙無法透過手術改善。手術目的在於矯正第一眼位偏位、減輕代償性頭位及改善誘發性眼瞼下垂 ,不預期能正常化眼球運動範圍。許多患者術後仍能藉由代償功能維持良好的雙眼視覺。
目前主流為神經源性理論。
發生時期 :胚胎發育4~8週時發生外展神經運動神經元缺損 。約翰霍普金斯大學的屍體解剖研究(1980年代)確立了神經源性機制。異常支配的機制 :由於外展神經缺損 ,動眼神經(CN3)的內收支異常支配外直肌。內收時內外直肌同時收縮,導致眼球後退。眼瞼裂狹小是因眼球後退而被動產生。EMG所見 :外展時外直肌電活動缺失,內收時出現矛盾性激活(1956年首次報告)已被證實。MRI所見 :可顯示外展神經缺損 或發育不良。I型80%缺損 ,II型則大多存在。DRS 是異常支配表現型的連續體(continuum)空間鄰近性 :海綿竇 及眼眶 尖端內CN3與CN6的空間鄰近性促進軸突錯誤導向。肌源性理論(歷史) :曾認為外直肌纖維化或內直肌異常後方附著是原因,但無法充分解釋矛盾性運動模式,目前神經源性理論為主流。
作為原發性軸突缺陷,CN3對外直肌的異常支配並非CN3本身的發育異常(原發性),而是CN6缺損 導致外直肌未被支配,進而吸引CN3軸突的繼發性現象,此點已在動物模型中證實2) 。MafB是在菱腦節5-6表達的轉錄因子,發育中的動眼神經運動神經元不表達。因此,CN3向外直肌的異常分支並非CN3的細胞自主性變化,而是CN6支配缺失所致的繼發性現象2) 。
CHN1基因敲入小鼠會發生外展神經軸突停滯,隨後導致外展運動神經元消失。CHN1突變涉及α2-chimaerin作為EphA4下游效應器,並參與介導ephrin-A5排斥信號的路徑2) 。
Al-Dabet S, et al. Abnormal head position in strabismus: a comprehensive review. Surv Ophthalmol. 2025;70:771-816.
Whitman MC, Engle EC. Axon abnormalities in congenital cranial dysinnervation disorders. Annu Rev Vis Sci. 2020;6:51-76.
開啟下方的 AI 助手,並將複製的內容貼到聊天欄。