杜安综合征(DRS )是一种非进行性斜视 综合征,本质为外展神经(CN6)先天性缺失和动眼神经对外直肌的异常支配。
是最常见的先天性脑神经异常支配疾病(CCDD ),在一般人群中约每1000人中有1人发生2) 。
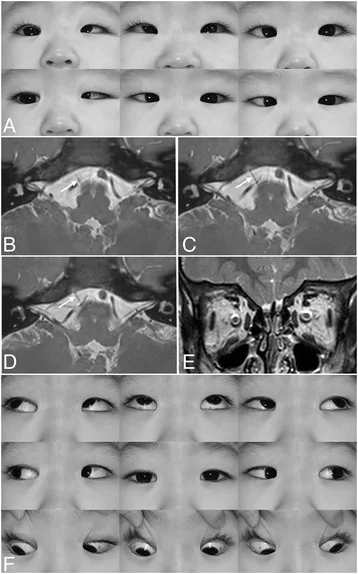
内转时眼球后退和睑裂缩小是极为特征性的表现。
I型(外转受限为主)最常见,约占全部病例的85%。多数在10岁前确诊。
大多数患者双眼视功能正常,通过代偿性头位(面转)维持双眼视,视力 障碍较少。
手术并非眼球运动障碍 的根本治疗,目的是矫正第一眼位偏斜和改善头位。
杜安综合征(Duane Syndrome; Duane Retraction Syndrome, DRS )是1905年由Alexander Duane详细描述的先天性、非进行性斜视 综合征。也称为Stilling-Türk-Duane综合征 ,ICD-10编码为H50.81。是一种先天性眼球运动障碍 ,特征为严重的外转障碍以及内转时的眼球后退和由此引起的睑裂缩小。
一般人群中约每1000人中有1人发病2) ,占所有斜视 病例的4%。是先天性颅神经异常支配疾病(CCDD )中最常见的疾病2) 。单侧占82%,多见于左眼(59%),男女比为4:6,女性略多。双侧占15-20%。多数在10岁前确诊。
I型(约85%)
特征 :外展受限 > 内收受限。正位视可呈现内斜视 。
头位 :最多见为向患侧转脸。72%有面部转向(68%转向患侧)1)
MRI所见 :约80%外展神经缺失。
流行病学 :Anvari(125例)中I型占87.0%1)
II型及III型
II型(5〜10%) :内收受限 > 外展受限。正位视呈现外斜视 ,面部转向与I型相反。MRI显示大多数病例外展神经存在。
III型(10〜20%) :内外展均受限。93%有面部转向(73%转向患侧对侧)1)
大样本研究(441例)中,面部旋转占54.6%,单侧病例中面部旋转的发生率显著更高1) 。水平偏位占76.0%,内斜视 占58.4%,外斜视 占17.6%1) 。
Q
杜安综合征是一种多常见的疾病?
A
一般人群中约每1000人中有1人发病2) ,占所有斜视 的4%。在先天性颅神经异常支配疾病(CCDD )中最为常见。单侧占82%,左眼和女性略多见。多数在10岁前确诊。
Kim JH, et al. Postoperative full abduction in a patient of Duane retraction syndrome without an abducens nerve: a case report. BMC Ophthalmol. 2017. Figure 1. PMCID: PMC5438545. License: CC BY.
左杜安综合征多方向注视:左眼外展受限、内转时眼裂缩小、轻度上射。对应本文“主要症状与临床所见”部分讨论的特征性眼球运动异常。
双眼视功能 :大多数病例双眼视功能正常,很少引起视力 障碍。代偿性头位旋转 :为克服外展障碍,通过向患侧旋转面部来维持双眼视。需要向家长和教师充分说明,这种头位异常是为了代偿眼球运动障碍 、实现双眼视所必需的动作。弱视 弱视 发生率为5.1%1) 。屈光 不正远视 最多。据报道远视 42.3%、近视 39.7%1) 。由于常合并屈光 不正,可能需要治疗屈光参差性弱视 。
外展受限 :完全或部分外展受限(程度因类型而异)。眼球后退+睑裂变窄 :内转时眼球后退,睑裂变窄(诱发性上睑下垂 )。这是DRS 的特征性表现,从正侧面观察容易确认。当内转时睑裂变窄不明显时,从正侧面观察眼球的同时使其内转,则眼球后退更易识别。睑裂增宽 :外展时睑裂增宽。上射/下射(牵拉现象) :内转时眼球向上或向下偏移。出生时并不出现,而是由于外直肌进行性挛缩继发产生。内转时外直肌向上偏移则出现上射,向下偏移则出现下射。辐辏不全 :可能伴有辐辏反射减弱。
30%~50%的患者存在先天性脑干障碍引起的异常。具体例子包括鳄鱼泪(味觉性流泪)和感音神经性耳聋。
相关综合征包括Duane radial ray综合征(SALL4突变)、Goldenhar综合征、HOXA1综合征等2) 。
Q
代偿性面部旋转需要治疗吗?
A
代偿性头位旋转是为了维持双眼视功能的适应动作。轻度(小于15°)原则上无需治疗,仅需观察。当旋转角度大于15°或引起日常生活及美观问题时,考虑手术。在学校,可将儿童安排在面向旋转方向的位置,环境调整也有效。
外展神经(CN6)缺失 :胚胎发育4~8周时受损,导致外展神经运动神经元缺失或发育不良。患侧脑干的外展神经核及外展神经发育不良或缺失。动眼神经(CN3)异常支配 :CN3内转支异常支配外直肌,导致内转时内直肌和外直肌同时收缩,引起眼球后退。睑裂狭小是由眼球后退被动产生的。外直肌的变化 :组织学上,外直肌的一部分(未接受正常神经支配的部分)发生纤维化。CCDD 之一2) 。
孤立型中10%为遗传性,90%为散发性。I型为常染色体显性遗传 (8q13),II型为常染色体显性遗传 (DURS2: CHN1突变, 2q31-q32.1)。主要基因突变如下2) 。
CHN1突变 :α2-chimaerin(Rac-GAP蛋白)的错义突变。破坏蛋白质闭合结构的维持残基,导致RacGAP活性过度增强。CHN1突变病例多为双侧DRS ,可伴有垂直方向异常。MAFB突变 :杂合性功能丧失突变导致散发性DRS 。部分病例表现为DRS +听力丧失+局灶节段性肾小球硬化症(FSGS)。SALL4突变 :Duane radial ray综合征(DRS +上肢畸形),常染色体显性遗传 。HOXA1突变 :双侧DRS +感音神经性耳聋+面神经麻痹+中枢性低通气+血管畸形+智力障碍。COL25A1突变 :先天性上睑下垂 或DRS (同一家族内表型不同)。
遵循标准斜视 评估(视力 、眼球运动、双眼视、屈光 检查)。
裂隙灯 及眼球运动检查 DRS 的特征。从侧面观察更容易确认眼球后退。眼球牵拉试验 :阳性(中枢性但外眼肌有挛缩性改变)。有助于鉴别机械性限制和神经支配缺陷。MRI :可显示外展神经缺失或发育不良。I型80%的病例MRI显示外展神经缺失,而II型大多数外展神经存在。常规诊断不推荐,但对术前评估有用。基因检测(CHN1) :仅推荐用于家族性病例。
疾病 鉴别要点 先天性外展神经麻痹 无眼球后退,内斜视 角较大 HGPPS 水平注视麻痹+进行性脊柱侧弯,无眼球后退2) Moebius综合征 CN6+CN7障碍,伴有面部肌力低下2) Brown综合征 以上转受限为主,无眼球后退
许多病例无需治疗。眼球运动障碍 本身没有根本性治疗方法。
屈光 矫正(眼镜)远视 即可消除面部转动和内斜视 的病例存在1) 。优先治疗屈光 不正和屈光参差性弱视 。棱镜眼镜 弱视 治疗弱视 时进行标准遮盖疗法 。多数Duane综合征 患者通过面部转动实现双眼视,因此斜视 性弱视 的风险不高。肉毒杆菌毒素注射 1) 。Ameri的16名研究中,面部旋转18.27°±7.29°在1周后改善至0.094°,但6个月后复发至7°1) 。
手术适应证 :异常头位≥15°,第一眼位明显偏斜,重度诱发性上睑下垂 (睑裂宽度减少≥50%)。
内斜视DRS
内直肌后徙术 是基础。适用于I型Duane综合征 伴内斜视 或明显异常头位者。
内斜视 <15Δ时,单眼内直肌后徙6mm 可矫正。超过此度数需行双眼内直肌后徙。
注意:后徙超过5mm会增加内转受限和继发性外斜视 的风险。
Pressman的19名患者(术前26.28PD→术后2.71PD,成功率79%)1)
有报告称,西田法联合内直肌后徙术可将40PD的内斜视 改善至6PD,外展恢复至45°1) 。
重症及难治病例
对于上/下转偏斜 ,可行外直肌后徙术或外直肌Y形劈开术。无论如何均无法根治,因此需谨慎确定适应证。
外直肌巩膜固定术 (或大量后徙)联合内直肌后徙术也有效。
垂直直肌移位术(VRT) :将上、下直肌移位至外直肌附着点。增强缝合组AHP从22.7°改善至3.6°,非增强组从18.7°改善至7°1) 。
手术并非DRS 的根本性治疗。术后神经支配缺陷仍持续存在。
术前需说明手术目的是矫正第一眼位偏斜、改善头位、减轻诱发性上睑下垂 。
内直肌过度后徙有内转受限、继发性外斜视 的风险。
Q
肉毒杆菌毒素注射的有效程度如何?
A
肉毒杆菌毒素可暂时改善异常头位和内斜视 ,但效果在6个月后减弱1) 。作为手术的替代或辅助疗法,可考虑用于婴幼儿早期干预或手术风险高的病例。有时需要重复注射。
Q
手术能治愈杜安综合征吗?
A
外展神经先天性缺失和动眼神经异常支配这一根本性神经障碍无法通过手术改善。手术的目的是矫正第一眼位偏位、减轻代偿性头位、改善诱发性上睑下垂 ,不期望眼球运动范围恢复正常。许多患者术后仍能通过代偿功能维持良好的双眼视。
目前主流理论是神经源性理论。
发生时期 :胚胎发育4~8周时外展神经运动神经元缺失。约翰·霍普金斯大学的尸检研究(1980年代)确立了神经源性机制。异常支配的机制 :由于外展神经缺损 ,动眼神经(CN3)的内直肌支异常支配外直肌。内转时内直肌和外直肌同时收缩,导致眼球后退。睑裂缩小是由于眼球后退被动产生的。EMG表现 :外转时外直肌电活动缺失,内转时出现反常激活(1956年首次报道)已被证实。MRI表现 :可显示外展神经缺损 或发育不良。I型中80%存在缺损 ,II型中几乎都存在。DRS 是异常支配表现型的连续谱空间邻近性 :海绵窦 和眶尖内CN3与CN6的空间邻近性促进了轴突的错误导向。肌源性理论(历史) :曾有一段时间认为外直肌纤维化或内直肌异常后附着是原因,但无法充分解释反常运动模式,目前神经源性理论是主流。
作为原发性轴突缺陷,CN3对外直肌的异常支配并非CN3本身发育异常(原发性),而是由于CN6缺损 导致外直肌未被支配,从而吸引CN3轴突的继发性现象,这在动物模型中已得到证实2) 。MafB是在菱脑节5-6中表达的转录因子,在发育中的动眼神经运动神经元中不表达。因此,CN3向外直肌的异常分支不是CN3细胞自主性变化,而是由于CN6支配缺失引起的继发性现象2) 。
CHN1敲入小鼠会出现外展神经轴突停滞,随后外展运动神经元消失。CHN1突变涉及α2-chimaerin作为EphA4下游效应器,介导ephrin-A5排斥信号的通路2) 。
Al-Dabet S, et al. Abnormal head position in strabismus: a comprehensive review. Surv Ophthalmol. 2025;70:771-816.
Whitman MC, Engle EC. Axon abnormalities in congenital cranial dysinnervation disorders. Annu Rev Vis Sci. 2020;6:51-76.
复制全文后,可以粘贴到你常用的 AI 助手中提问。
打开下面的 AI 助手,并把复制的内容粘贴到聊天框。