El síndrome de córnea frágil (BCS; OMIM 229200, 614170) es un trastorno del tejido conectivo autosómico recesivo raro caracterizado por adelgazamiento corneal progresivo y esclera azul. Fue reportado por primera vez por Stein et al. en 19682). La prevalencia se estima en menos de 1 por cada 1,000,000 de personas1). Hasta 2021, se han reportado un total de 86 casos, con muchos casos con antecedentes familiares de matrimonio consanguíneo1).
El BCS se clasifica en dos tipos según el gen causante1). Ambos tipos codifican factores de transcripción involucrados en el mantenimiento de la homeostasis de la matriz extracelular1)2). La alteración en la deposición de colágeno y el ensamblaje de fibras conduce a la fragilidad estructural del estroma corneal1).
BCS tipo 1 (mutación ZNF469)
Gen causante: ZNF469 (16q24), un gen de un solo exón que codifica 3,953 aminoácidos1)
Número de pacientes reportados: 53 casos (24 mutaciones identificadas)1)
Principales tipos de mutación: Mayoritariamente mutaciones homocigotas de cambio de marco o sin sentido1)
Nota especial: Se ha informado que las mutaciones heterocigotas están asociadas con el queratocono2)
BCS tipo 2 (mutación PRDM5)
Gen causante: PRDM5 (4q27) es un gen de 16 exones que codifica 630 aminoácidos1)
Número de pacientes reportados: 33 casos (14 mutaciones diferentes identificadas)1)
Principales tipos de mutación: Todas reportadas como mutaciones homocigotas1)
Nota especial: También participa en el desarrollo de los microvasos retinianos y la membrana de Bruch1)
Q¿En qué se diferencia el BCS del síndrome de Ehlers-Danlos?
A
El BCS se consideraba anteriormente parte del síndrome de Ehlers-Danlos cifoescoliótico (EDS tipo VI). Sin embargo, el análisis genético molecular ha confirmado que es una enfermedad distinta1). El EDS VI es causado por una deficiencia de lisil hidroxilasa debido a mutaciones en el gen PLOD1. Se puede diferenciar por una elevación de la relación desoxipiridinolina/piridinolina en orina, que es normal en el BCS1). El EDS VI tiene un mal pronóstico debido a la rotura arterial, mientras que la esperanza de vida en el BCS se considera normal.
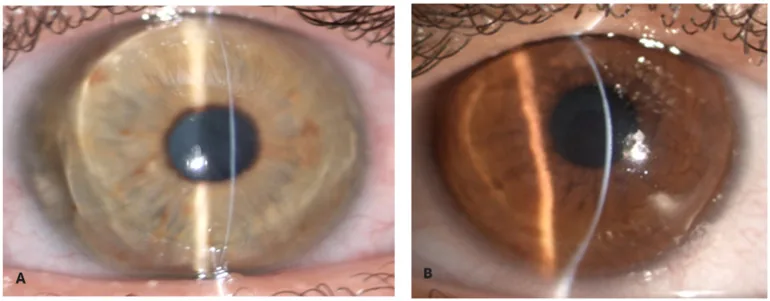
Zeppieri M, Nabil R, Ruzza A, et al. Brittle Cornea Syndrome: Molecular Diagnosis and Management. Diagnostics (Basel). 2025 Jun 24;15(13):1596. Figure 1. PMCID: PMC12249002. License: CC BY.
Con una hendidura estrecha de la lámpara de hendidura, el estroma corneal aparece extremadamente delgado y abombado hacia adelante. El adelgazamiento extremo y la protrusión cónica muestran la apariencia de una córnea frágil.
La miopía progresiva y la pérdida de visión debido al astigmatismo irregular son las principales quejas3). En el momento de la perforación corneal, se produce dolor repentino y pérdida de visión5). También puede notarse pérdida auditiva debido a sordera1).
El adelgazamiento de toda la córnea (de limbo a limbo) es lo más característico. El grosor corneal central (CCT) suele ser inferior a 400 μm1). En un informe de tres hermanos, el CCT fue de 243 a 304 μm5). En hermanos albaneses, fue de 189 μm y 157 μm3). En dos hermanos de Nueva Zelanda, se confirmó un adelgazamiento extremo de 167 μm y 149 μm4).
La perforación corneal ocurre a una edad promedio de 4.3 años (rango 1.5–19 años) 1). Se observa perforación ocular en más de dos tercios de los casos reportados 1). En más de la mitad de los casos se produce pérdida permanente de la visión 1). La esclerótica azul es el hallazgo ocular más frecuente, observado en 72 de 78 casos 1).
La hipermovilidad articular es el hallazgo extraocular más frecuente 1). También se han reportado displasia congénita de cadera, escoliosis y pie plano 1). La pérdida auditiva tiende a afectar más gravemente las frecuencias altas 1). La hipercomplacencia de la membrana timpánica es característica 1). La piel es blanda, hiperextensible y propensa a hematomas fáciles 1).
Los informes de fragilidad ósea también han aumentado en los últimos años. Dos hermanos con mutaciones heterocigotas compuestas en ZNF469 presentaron más de 10 fracturas y osteopenia 4). Se ha sugerido que aproximadamente el 16% de los pacientes con BCS pueden tener fragilidad ósea 4). La biopsia ósea mostró adelgazamiento del hueso cortical y una marcada disminución de la porosidad cortical 4).
El BCS es un trastorno autosómico recesivo causado por mutaciones bialélicas en dos genes 1).
ZNF469 participa en el desarrollo normal de la cámara anterior y la córnea1). Los estudios de asociación del genoma completo han mostrado una asociación con el grosor corneal central 4). PRDM5 regula directamente la transcripción de genes de colágeno fibrilar 4). Las mutaciones de pérdida de función (mutaciones de cambio de marco y sin sentido) representan una alta proporción en ambos genes 1).
Las mutaciones heterocigotas en ZNF469 se asocian con una reducción del grosor corneal central 2). Sin embargo, no siempre conducen a un adelgazamiento corneal. Un varón chino de 64 años con una mutación heterocigota en ZNF469 presentaba un grosor corneal normal (aproximadamente 550 μm), pero mostró opacidad corneal y defectos epiteliales 2).
El matrimonio consanguíneo es un factor de riesgo importante, ya que aumenta la probabilidad de mutaciones homocigotas 1). En tres casos turcos se identificó una mutación común PRDM5 c.17T>G, p.(Val6Gly), lo que sugiere una posible mutación fundadora 1).
El diagnóstico clínico se basa en el adelgazamiento generalizado de la córnea (grosor corneal central inferior a 400 μm), esclerótica azul y síntomas sistémicos como hipermovilidad articular y pérdida auditiva 3). La paquimetría corneal y la topografía corneal mediante dispositivos como Pentacam son esenciales 3). La OCT del segmento anterior es útil para la evaluación detallada de la estructura corneal 2)3).
Para un diagnóstico definitivo son necesarias las pruebas genéticas de ZNF469 y PRDM5 3). La secuenciación del exoma completo (WES) es eficaz para identificar variantes patogénicas 3). También proporciona información útil para el asesoramiento genético y la planificación familiar 3). Los portadores pueden desarrollar miopía y adelgazamiento corneal leve 2).
Disección aórtica, estatura alta, luxación del cristalino
En BCS, la diferenciación del EDS cifoescoliótico (kEDS-PLOD1) es particularmente importante. En kEDS-PLOD1, cuando ocurre ruptura ocular, la esclerótica es más propensa a romperse que la córnea1). En kEDS-PLOD1, la escoliosis, la hipotonía y las complicaciones vasculares son más prominentes1).
Q¿Por qué es importante el diagnóstico temprano de BCS?
A
La edad promedio de inicio de la perforación corneal es de 4.3 años, y la reparación quirúrgica después de la perforación es extremadamente difícil5). Más de la mitad de los casos reportados resultan en pérdida permanente de la visión1). El diagnóstico temprano permite el uso de gafas protectoras y educación para la prevención de traumatismos1). El cribado de hermanos también es importante para la detección temprana5).
El aspecto más importante del manejo de BCS es la prevención de la perforación corneal1). Se recomienda el uso continuo de gafas protectoras de policarbonato5). También se puede considerar la administración profiláctica de medicamentos para reducir la presión intraocular. Las lentes de contacto están restringidas debido al adelgazamiento corneal y al riesgo de traumatismo1).
Indicaciones: Se realiza para la recuperación visual en casos de adelgazamiento corneal severo o después de una perforación3)
Técnica quirúrgica: uso de trépano de 8.0 mm, 16 suturas interrumpidas de nailon 10-0 3)
Resultados: la mejor agudeza visual corregida (MAVC) mejoró de 20/200 a 20/30 y 20/25 en hermanos albaneses 3)
Evolución: sin complicaciones durante 7 años 3)
Queratoplastia lamelar anterior profunda (DALK)
Ventajas: preservar el endotelio reduce el riesgo de rechazo 3)
Limitaciones: riesgo de perforación intraoperatoria debido a la fragilidad del estroma 3)
Caso reportado: en un caso se produjo perforación endotelial central intraoperatoria y se convirtió a PKP3)
Criterios de selección: se requiere un grosor estromal adecuado e integridad de la membrana de Descemet3)
En la reparación de perforaciones corneales, la fragilidad extrema del tejido es un problema. En un informe de tres hermanos, el uso de puntos de sutura largos, adhesivo de cianoacrilato y lente de contacto de vendaje fue efectivo 5). La creación cuidadosa de un túnel corneoescleral puede reducir las complicaciones 5). Un paciente de 64 años con BCS se sometió a PKP y tuvo una agudeza visual corregida de 0.2 al alta 2).
Q¿Es efectivo el cross-linking de colágeno corneal?
A
Dos pacientes pediátricos con BCS con grosor corneal central inferior a 280 μm se sometieron a CXL transepitelial (dosis de UV ajustada según el grosor corneal) 3). Se informó mejoría en la agudeza visual y mantenimiento de la densidad de células endoteliales 3). Sin embargo, el protocolo estándar de Dresde requiere un grosor corneal central de al menos 400 μm y está contraindicado en córneas ultrafinas de menos de 200 μm 3). Se espera que las modificaciones del protocolo amplíen las indicaciones, pero actualmente es limitado.
Tanto ZNF469 como PRDM5 codifican factores de transcripción 1). ZNF469 tiene tres dominios de dedos de zinc tipo C2H2 en el extremo C 1). Regula la expresión de genes de la matriz extracelular (CLU, GPC6, PCOLCE2, THBS1) 1). PRDM5 tiene un dominio PR SET y regula directamente la transcripción de genes de colágeno mediante la unión a la ARN polimerasa II 4). También participa en la regulación de la vía de señalización Wnt.
Estas mutaciones genéticas alteran el depósito de colágeno y el ensamblaje de fibrillas en el estroma corneal1). Las mutaciones en ZNF469 causan una expresión reducida y cambios estructurales del colágeno tipo I (COL-I) 2). La inmunofluorescencia del tejido corneal después de QP mostró disminución de COL-I y aumento de colágeno tipo III 2). La tinción de Masson también demostró una clara reducción en la cantidad de fibras de colágeno 2).
La microscopía confocal revela estrías reticulares hiperreflectivas en el estroma corneal anterior 2). No se acompaña de infiltración de células inflamatorias 2). Esto sugiere que las lesiones corneales en BCS se deben a una anomalía primaria en la estructura del colágeno.
En el tejido óseo, PRDM5 también se une al ADN exónico de los genes de colágeno tipo I 4). La biopsia ósea mostró adelgazamiento de la cortical y una marcada reducción de la porosidad cortical (1.3%; normal 6.3±0.6%) 4). Un panel de 28 genes de fragilidad ósea no detectó otras mutaciones patogénicas, lo que sugiere que las mutaciones en ZNF469 pueden ser la causa de la fragilidad ósea 4).
Q¿Por qué se afectan otros tejidos además de la córnea en BCS?
A
ZNF469 y PRDM5 participan en la regulación transcripcional de la matriz extracelular en los tejidos conectivos de todo el cuerpo 1). Las anomalías estructurales de las fibras de colágeno no se limitan a la córnea, sino que afectan a los tejidos conectivos sistémicos, incluyendo la esclerótica (coloración azul), articulaciones (hipermovilidad), piel (hiperextensibilidad), hueso (osteopenia) y membrana timpánica (hipercomplacencia) 4). Se han reportado osteopenia y fracturas, lo que sugiere que BCS puede tener un fenotipo de fragilidad ósea 4).
Los avances en terapia génica están abriendo nuevas posibilidades para BCS. La edición genómica CRISPR y la interferencia de ARN (ARNi) se esperan como tratamientos destinados a estabilizar o mejorar el adelgazamiento corneal 3).
Los implantes corneales bioingenieriles (BPCDX) son hidrogeles transparentes acelulares hechos de colágeno porcino de grado médico 3). Se insertan en un bolsillo intraestromal de 2–3 mm creado con láser de femtosegundo, un procedimiento mínimamente invasivo 3). A los 24 meses, se ha reportado aumento del grosor corneal, aplanamiento de la queratometría máxima en 18 D y mejora de la agudeza visual corregida3). Se han obtenido resultados estables sin rechazo 3).
También se está estudiando la inyección intraestromal de células madre mesenquimales 3). Las células madre derivadas de tejido adiposo y médula ósea se inyectan en una disección lamelar creada con láser de femtosegundo, lo que podría retrasar la necesidad de QP 3). Actualmente solo se usa en pacientes con queratocono, pero se espera su aplicación en pacientes con BCS 3).
Sanri A, Demir S, Gürkan H. Homozygous Val6Gly Variation in PRDM5 Gene Causing Brittle Cornea Syndrome: A New Turkish Case. Mol Syndromol. 2023;14(2):129-135. doi:10.1159/000524832. PMID:37064337; PMCID:PMC10091010.
Geng X, Zhu L, Li J, Li Z. Brittle cornea syndrome: A novel mutation. Heliyon. 2024;10:e32506. doi:10.1016/j.heliyon.2024.e32506.
Zeppieri M, Gentile M, Acquaviva A, Scollo D, D’Esposito F, Gagliano G, et al. Brittle Cornea Syndrome: Molecular Diagnosis and Management. Diagnostics (Basel, Switzerland). 2025;15(13). doi:10.3390/diagnostics15131596. PMID:40647596; PMCID:PMC12249002.
Cundy T, Vincent A, Robertson S. Does brittle cornea syndrome have a bone fragility phenotype? Bone Rep. 2021;15:101124. doi:10.1016/j.bonr.2021.101124. PMID:34522702; PMCID:PMC8426531.
Mandlik K, Betdur RA, Rashmita R, Narayana S. Brittle cornea syndrome: A tale of three brothers. Indian journal of ophthalmology. 2022;70(7):2594-2597. doi:10.4103/ijo.IJO_2894_21. PMID:35791165; PMCID:PMC9426125.
Copia el texto del artículo y pégalo en el asistente de IA que prefieras.
Artículo copiado al portapapeles
Abre un asistente de IA abajo y pega el texto copiado en el chat.