BCS 1型(ZNF469突变)

脆性角膜综合征
一目了然的要点
Section titled “一目了然的要点”1. 什么是脆性角膜综合征?
Section titled “1. 什么是脆性角膜综合征?”脆性角膜综合征(BCS;OMIM 229200, 614170)是一种罕见的常染色体隐性遗传性结缔组织疾病,以进行性角膜变薄和蓝色巩膜为特征。1968年由Stein等人首次报道2)。患病率估计低于百万分之一1)。截至2021年,累计报告86例,其中许多病例有近亲结婚的家族史1)。
BCS根据致病基因分为两型1)。两种类型均编码参与维持细胞外基质稳态的转录因子1)2)。胶原沉积和纤维组装的障碍导致角膜基质的结构脆弱性1)。
BCS 2型(PRDM5突变)
致病基因:PRDM5(4q27)是一个16外显子基因,编码630个氨基酸1)
报告患者数:33例(已鉴定出14种突变)1)
主要突变类型:所有报道均为纯合突变1)
特别说明:也参与视网膜微血管和布鲁赫膜的发育1)
Q
BCS与埃勒斯-丹洛斯综合征有何不同?
A
BCS以前被认为是脊柱后侧凸型埃勒斯-丹洛斯综合征(EDS VI型)的一部分。然而,分子遗传学分析已证实它是一种独立的疾病1)。EDS VI由PLOD1基因突变导致的赖氨酰羟化酶缺乏引起。可通过尿中脱氧吡啶啉/吡啶啉比值升高来鉴别,而BCS中该比值正常1)。EDS VI因动脉破裂预后不良,而BCS的预期寿命被认为是正常的。
2. 主要症状和临床所见
Section titled “2. 主要症状和临床所见”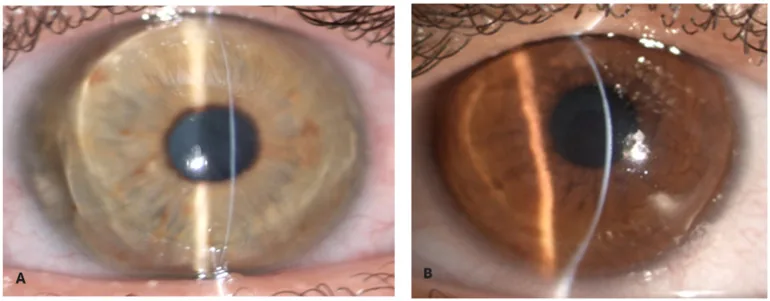
Zeppieri M, Nabil R, Ruzza A, et al. Brittle Cornea Syndrome: Molecular Diagnosis and Management. Diagnostics (Basel). 2025 Jun 24;15(13):1596. Figure 1. PMCID: PMC12249002. License: CC BY.
进行性近视和不规则散光导致的视力下降是主要主诉3)。角膜穿孔时会出现突然的疼痛和视力丧失5)。也可能自觉听力下降导致的听觉障碍1)。
整个角膜(从角膜缘到角膜缘)变薄是最具特征性的表现。中央角膜厚度(CCT)通常小于400 μm1)。在一份三兄弟的报告中,CCT为243–304 μm5)。在阿尔巴尼亚裔姐弟中,分别为189 μm和157 μm3)。在新西兰的两兄弟中,确认了极度的变薄,分别为167 μm和149 μm4)。
角膜穿孔平均发病年龄为4.3岁(范围1.5-19岁)1)。超过三分之二的报告病例出现眼球穿孔1)。半数以上病例发生永久性视力丧失1)。蓝色巩膜是最常见的眼部表现,78例中有72例出现1)。
| 系统 | 主要表现 | 频率 |
|---|---|---|
| 眼部 | 角膜变薄/穿孔、蓝色巩膜 | >90% 1) |
| 关节 | 小关节过度活动 | 64/78例 1) |
| 听觉 | 感音神经性、传导性或混合性听力损失 | 32/78例 1) |
关节过度活动是最常见的眼外表现1)。先天性髋关节发育不良、脊柱侧弯和扁平足也有报道1)。听力损失倾向于更严重地影响高频1)。鼓膜过度顺应性是其特征1)。皮肤柔软、过度伸展且易出现瘀伤1)。
近年来骨脆性的报告也有所增加。两名携带ZNF469复合杂合突变的兄弟有超过10次骨折和骨量减少4)。提示约16%的BCS患者可能存在骨脆性4)。骨活检显示皮质骨变薄和皮质骨孔隙率显著降低4)。
3. 病因与风险因素
Section titled “3. 病因与风险因素”BCS是一种常染色体隐性遗传病,由两个基因的双等位基因突变引起1)。
ZNF469参与前房和角膜的正常发育1)。全基因组关联分析显示其与中央角膜厚度相关4)。PRDM5直接调节纤维胶原基因的转录4)。两个基因的功能丧失型突变(移码突变和无义突变)占比较高1)。
ZNF469的杂合突变与中央角膜厚度减少相关2),但并不一定导致角膜变薄。一例64岁中国男性携带ZNF469杂合突变,角膜厚度正常(约550μm),但出现角膜混浊和上皮缺损2)。
近亲结婚是重要的风险因素,会增加纯合突变的发生概率1)。在三例土耳其病例中,发现共同的PRDM5 c.17T>G, p.(Val6Gly)突变,提示可能存在创始者突变1)。
4. 诊断与检查方法
Section titled “4. 诊断与检查方法”临床诊断基于角膜整体变薄(中央角膜厚度小于400μm)、蓝色巩膜以及关节过度活动、听力丧失等全身症状3)。角膜厚度测量和Pentacam等角膜地形图检查必不可少3)。前段OCT有助于详细评估角膜结构2)3)。
确诊需要对ZNF469和PRDM5进行基因检测3)。全外显子组测序(WES)可有效识别致病性变异3)。基因检测也为遗传咨询和计划生育提供有用信息3)。携带者可能出现近视和轻度角膜变薄2)。
| 鉴别疾病 | 与BCS的区别 |
|---|---|
| 脊柱后侧凸型EDS | 动脉破裂风险、尿赖氨酰吡啶啉比值升高1) |
| 成骨不全症 | 反复骨折为主要特征、牙发育不全4) |
| 马凡综合征 | 主动脉夹层、身材高大、晶状体脱位 |
在BCS中,与脊柱后侧凸型EDS(kEDS-PLOD1)的鉴别尤为重要。kEDS-PLOD1发生眼球破裂时,巩膜比角膜更容易破裂1)。kEDS-PLOD1中脊柱侧弯、肌张力低下和血管并发症更为显著1)。
Q
为什么BCS的早期诊断很重要?
A
角膜穿孔的平均发病年龄为4.3岁,穿孔后的外科修复极为困难5)。超过一半的报告病例导致永久性视力丧失1)。早期诊断可以采取佩戴防护眼镜和外伤预防教育1)。兄弟姐妹的筛查对早期发现也很重要5)。
5. 标准治疗方法
Section titled “5. 标准治疗方法”BCS管理的最重要项目是预防角膜穿孔1)。建议持续佩戴聚碳酸酯防护眼镜5)。也可考虑预防性使用降眼压药物。由于角膜变薄和外伤风险,限制使用隐形眼镜1)。
穿透性角膜移植术(PKP)
适应症:用于严重角膜变薄或穿孔后的视力恢复3)
手术方式:使用8.0mm环钻,16根10-0尼龙线间断缝合3)
结果:阿尔巴尼亚裔兄妹的最佳矫正视力(BCVA)从20/200改善至20/30和20/253)
经过:7年内无并发症3)
深板层角膜移植术(DALK)
角膜穿孔修复中,组织极度脆弱是一个问题。三兄弟的报告显示,使用长缝线咬合、氰基丙烯酸酯粘合剂和绷带式隐形眼镜有效5)。仔细制作角巩膜隧道可减少并发症5)。一名64岁的BCS患者接受了PKP,出院时矫正视力为0.22)。
Q
角膜胶原交联有效吗?
A
两名中央角膜厚度小于280μm的儿童BCS患者接受了经上皮CXL(根据角膜厚度调整紫外线照射剂量)3)。报告显示视力改善和内皮细胞密度维持3)。然而,标准德累斯顿方案要求中央角膜厚度至少400μm,对于小于200μm的超薄角膜禁忌使用3)。方案修改有望扩大适应症,但目前仍有限。
6. 病理生理学与详细发病机制
Section titled “6. 病理生理学与详细发病机制”ZNF469和PRDM5均编码转录因子1)。ZNF469在C端有三个C2H2型锌指结构域1)。它调节细胞外基质基因(CLU、GPC6、PCOLCE2、THBS1)的表达1)。PRDM5具有PR SET结构域,通过与RNA聚合酶II结合直接调节胶原基因的转录4)。它还参与Wnt信号通路的调节。
这些基因突变导致角膜基质中胶原沉积和纤维组装受损1)。ZNF469突变导致I型胶原(COL-I)表达降低和结构改变2)。PKP后角膜组织的免疫荧光染色显示COL-I减少和III型胶原增加2)。Masson染色也证实胶原纤维量明显减少2)。
共聚焦显微镜显示前部角膜基质中有高反射的网状条纹组织2)。无炎症细胞浸润2)。这表明BCS的角膜病变源于胶原结构的原发性异常。
在骨组织中,PRDM5也与I型胶原基因的外显子DNA结合4)。骨活检显示皮质骨变薄和皮质骨孔隙率显著降低(1.3%;正常值6.3±0.6%)4)。28基因骨脆弱性面板未检测到其他致病突变,提示ZNF469突变本身可能导致骨脆弱性4)。
Q
为什么BCS会影响角膜以外的组织?
A
ZNF469和PRDM5参与全身结缔组织细胞外基质的转录调控1)。胶原纤维的结构异常不仅限于角膜,还影响全身结缔组织,包括巩膜(蓝色变色)、关节(过度活动)、皮肤(过度伸展)、骨骼(骨量减少)和鼓膜(过度顺应)4)。有骨量减少和骨折的报道,提示BCS可能具有骨脆弱性表型4)。
7. 最新研究与未来展望
Section titled “7. 最新研究与未来展望”基因治疗的进步为BCS带来了新的可能性。CRISPR基因组编辑和RNA干扰(RNAi)有望成为稳定或改善角膜变薄的治疗方法3)。
生物工程角膜植入物(BPCDX)是由医用猪胶原制成的无细胞透明水凝胶3)。通过飞秒激光制作2-3毫米的基质内口袋,采用微创方法植入3)。24个月的观察显示角膜厚度增加、最大角膜曲率平坦化18D、矫正视力改善3)。无排斥反应,结果稳定3)。
间充质干细胞角膜基质内注射也在研究中3)。将脂肪来源和骨髓来源的干细胞注入飞秒激光制作的层状切开部位,可能推迟PKP的需要3)。目前仅用于圆锥角膜患者,但有望应用于BCS患者3)。
8. 参考文献
Section titled “8. 参考文献”- Sanri A, Demir S, Gürkan H. Homozygous Val6Gly Variation in PRDM5 Gene Causing Brittle Cornea Syndrome: A New Turkish Case. Mol Syndromol. 2023;14(2):129-135. doi:10.1159/000524832. PMID:37064337; PMCID:PMC10091010.
- Geng X, Zhu L, Li J, Li Z. Brittle cornea syndrome: A novel mutation. Heliyon. 2024;10:e32506. doi:10.1016/j.heliyon.2024.e32506.
- Zeppieri M, Gentile M, Acquaviva A, Scollo D, D’Esposito F, Gagliano G, et al. Brittle Cornea Syndrome: Molecular Diagnosis and Management. Diagnostics (Basel, Switzerland). 2025;15(13). doi:10.3390/diagnostics15131596. PMID:40647596; PMCID:PMC12249002.
- Cundy T, Vincent A, Robertson S. Does brittle cornea syndrome have a bone fragility phenotype? Bone Rep. 2021;15:101124. doi:10.1016/j.bonr.2021.101124. PMID:34522702; PMCID:PMC8426531.
- Mandlik K, Betdur RA, Rashmita R, Narayana S. Brittle cornea syndrome: A tale of three brothers. Indian journal of ophthalmology. 2022;70(7):2594-2597. doi:10.4103/ijo.IJO_2894_21. PMID:35791165; PMCID:PMC9426125.