Le syndrome de la cornée fragile (Brittle Cornea Syndrome : BCS ; OMIM 229200, 614170) est une maladie rare du tissu conjonctif à transmission autosomique récessive caractérisée par un amincissement cornéen progressif et une sclère bleue. Il a été rapporté pour la première fois par Stein et al. en 1968 2). La prévalence est estimée à moins d’une personne par million 1). En 2021, 86 cas cumulés ont été rapportés, avec une fréquence élevée de consanguinité familiale 1).
Le BCS est classé en deux types selon le gène causal 1). Les deux types codent pour des facteurs de transcription impliqués dans l’homéostasie de la matrice extracellulaire 1)2). Les anomalies de dépôt de collagène et d’assemblage des fibrilles entraînent une fragilité structurelle du stroma cornéen1).
BCS type 1 (mutation ZNF469)
Gène causal : ZNF469 (16q24), un gène à exon unique codant pour 3 953 acides aminés 1)
Nombre de patients rapportés : 53 (24 mutations identifiées) 1)
Principaux types de mutations : Mutations faux-sens homozygotes ou non-sens fréquentes 1)
Remarque spéciale : Les mutations hétérozygotes ont été associées au kératocône2)
BCS type 2 (mutation PRDM5)
Gène causal : PRDM5 (4q27), un gène de 16 exons codant pour 630 acides aminés1)
Nombre de patients rapportés : 33 (14 mutations identifiées)1)
Principaux types de mutations : Toutes rapportées comme mutations homozygotes1)
Remarque spéciale : Également impliqué dans le développement des microvaisseaux rétiniens et de la membrane de Bruch1)
QEn quoi le BCS diffère-t-il du syndrome d'Ehlers-Danlos ?
A
Le BCS était auparavant considéré comme faisant partie du syndrome d’Ehlers-Danlos cyphoscoliotique (EDS type VI). Cependant, des analyses de génétique moléculaire ont confirmé qu’il s’agit d’une maladie distincte1). L’EDS VI est causé par un déficit en lysyl hydroxylase dû à des mutations du gène PLOD1. Il peut être différencié par un rapport désoxypyridinoline/pyridinoline urinaire élevé, alors que ce rapport est normal dans le BCS1). L’EDS VI a un mauvais pronostic vital en raison de ruptures artérielles, tandis que l’espérance de vie dans le BCS est considérée comme normale.
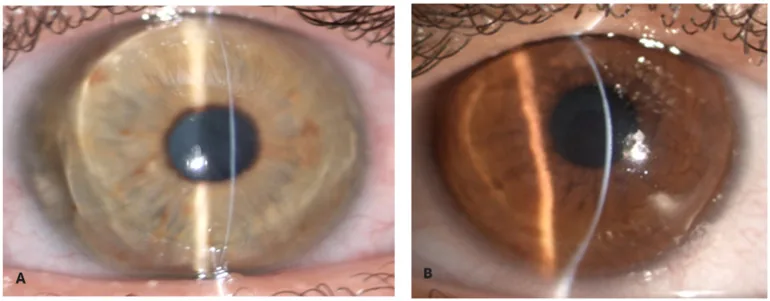
Zeppieri M, Nabil R, Ruzza A, et al. Brittle Cornea Syndrome: Molecular Diagnosis and Management. Diagnostics (Basel). 2025 Jun 24;15(13):1596. Figure 1. PMCID: PMC12249002. License: CC BY.
Avec une fente lumineuse étroite de la lampe à fente, le stroma cornéen apparaît extrêmement mince et bombé vers l’avant. L’amincissement extrême et la protrusion conique donnent l’apparence d’une cornée fragile.
La plainte principale est une baisse de l’acuité visuelle due à une myopie progressive ou à un astigmatisme irrégulier3). En cas de perforation cornéenne, une douleur soudaine et une perte de vision surviennent5). Une déficience auditive due à une surdité peut également être ressentie1).
L’amincissement de toute la cornée (de limbe à limbe) est le plus caractéristique. L’épaisseur cornéenne centrale (CCT) est souvent inférieure à 400 μm1). Dans un rapport sur trois frères, la CCT était de 243 à 304 μm5). Chez une fratrie albanaise, elle était de 189 μm et 157 μm3). Chez deux frères néo-zélandais, un amincissement extrême de 167 μm et 149 μm a été confirmé4).
La perforation cornéenne survient en moyenne à 4,3 ans (intervalle 1,5–19 ans) 1). Une perforation du globe oculaire est observée dans plus des deux tiers des cas rapportés 1). Une perte de vision permanente survient dans plus de la moitié des cas 1). La sclère bleue est le signe oculaire le plus fréquent, observé chez 72 des 78 cas 1).
Système
Principales constatations
Fréquence
Œil
Amincissement cornéen, perforation, sclère bleue
>90% 1)
Articulations
Hypermobilité des petites articulations
64/78 cas 1)
Audition
Surdité neurosensorielle, de transmission ou mixte
32/78 cas 1)
L’hypermobilité articulaire est la manifestation extraoculaire la plus fréquente 1). Une dysplasie congénitale de la hanche, une scoliose et des pieds plats ont également été rapportés 1). La perte auditive a tendance à affecter davantage les hautes fréquences 1). Une hypercomplaisance de la membrane tympanique est caractéristique 1). La peau est souple, hyperextensible et associée à une tendance aux ecchymoses 1).
Les rapports de fragilité osseuse ont également augmenté ces dernières années. Deux frères porteurs de mutations composites hétérozygotes de ZNF469 ont présenté plus de 10 fractures et une ostéopénie 4). Il a été suggéré qu’environ 16 % des patients atteints de BCS pourraient présenter une fragilité osseuse 4). La biopsie osseuse a montré un amincissement de l’os cortical et une diminution marquée de la porosité corticale 4).
Le BCS est une maladie autosomique récessive causée par des mutations bialléliques de deux gènes1).
ZNF469 est impliqué dans le développement normal de la chambre antérieure et de la cornée1). Les études d’association pangénomique ont montré une association avec l’épaisseur cornéenne centrale4). PRDM5 régule directement la transcription des gènes du collagène fibrillaire4). Les mutations avec perte de fonction (décalage du cadre de lecture, non-sens) sont fréquentes dans les deux gènes1).
Les mutations hétérozygotes de ZNF469 sont associées à une diminution de l’épaisseur cornéenne centrale2). Cependant, elles n’entraînent pas toujours un amincissement cornéen. Chez un homme chinois de 64 ans, une mutation hétérozygote de ZNF469 a montré une épaisseur cornéenne normale (environ 550 μm), mais une opacité cornéenne et une perte épithéliale ont été observées2).
La consanguinité est un facteur de risque important, augmentant la probabilité de mutations homozygotes1). Dans trois cas turcs, la mutation PRDM5 c.17T>G, p.(Val6Gly) était commune, suggérant un possible effet fondateur1).
Le diagnostic clinique repose sur un amincissement cornéen global (épaisseur cornéenne centrale < 400 μm), une sclère bleue, ainsi que des symptômes systémiques tels qu’une hypermobilité articulaire et une perte auditive3). La pachymétrie cornéenne et l’analyse de la morphologie cornéenne par Pentacam sont indispensables3). L’OCT du segment antérieur est utile pour une évaluation détaillée de la structure cornéenne2)3).
Le diagnostic définitif nécessite des tests génétiques pour ZNF469 et PRDM53). Le séquençage de l’exome entier (WES) est efficace pour identifier les variants pathogènes3). Il fournit également des informations utiles pour le conseil génétique et la planification familiale3). Les porteurs peuvent développer une myopie ou un léger amincissement cornéen2).
Dissection aortique, grande taille, luxation du cristallin
Dans le BCS, la distinction avec l’EDS cyphoscoliotique (kEDS-PLOD1) est particulièrement importante. Dans le kEDS-PLOD1, si une rupture oculaire survient, la sclère est plus susceptible de se rompre que la cornée1). Dans le kEDS-PLOD1, la scoliose, l’hypotonie musculaire et les complications vasculaires sont plus marquées1).
QPourquoi le diagnostic précoce du BCS est-il important ?
A
L’âge moyen d’apparition de la perforation cornéenne est de 4,3 ans, et la réparation chirurgicale après perforation est extrêmement difficile5). Plus de la moitié des cas rapportés entraînent une perte de vision permanente1). Un diagnostic précoce permet de porter des lunettes de protection et de mettre en place une éducation à la prévention des traumatismes1). Le dépistage des frères et sœurs est également important pour une détection précoce5).
L’élément le plus important de la prise en charge du BCS est la prévention de la perforation cornéenne1). Le port constant de lunettes de protection en polycarbonate est recommandé5). L’administration préventive de médicaments hypotenseurs oculaires est également envisagée. Les lentilles de contact sont limitées en raison de l’amincissement cornéen et du risque de traumatisme1).
Indication : Réalisée pour un amincissement cornéen sévère ou une récupération visuelle après perforation3)
Technique chirurgicale : utilisation d’un trépan de 8,0 mm, 16 sutures interrompues en nylon 10-03)
Résultats : chez une fratrie albanaise, la meilleure acuité visuelle corrigée (MAVC) est passée de 20/200 à 20/30 et 20/253)
Évolution : aucune complication pendant 7 ans3)
Kératoplastie lamellaire profonde (DALK)
Avantages : préserve l’endothélium, réduisant le risque de rejet3)
Limites : risque de perforation peropératoire en raison de la fragilité du stroma3)
Cas rapporté : dans un cas, perforation endothéliale centrale peropératoire ayant nécessité une conversion en PKP3)
Critères de sélection : épaisseur stromale suffisante et intégrité de la membrane de Descemet nécessaires3)
Dans la réparation des perforations cornéennes, la fragilité extrême des tissus pose problème. Dans un rapport sur trois frères, l’utilisation de points de suture longs, de colle cyanoacrylate et de lentilles de contact pansement a été efficace5). La création prudente d’un tunnel cornéoscléral peut réduire les complications5). Chez un patient BCS de 64 ans, une PKP a été réalisée et l’acuité visuelle corrigée à la sortie était de 0,22).
QLe cross-linking du collagène cornéen est-il efficace ?
A
Deux patients BCS pédiatriques avec une épaisseur cornéenne centrale < 280 μm ont bénéficié d’un CXL transépithélial (avec ajustement de la dose UV selon l’épaisseur cornéenne)3). Une amélioration de l’acuité visuelle et le maintien de la densité cellules endothéliales ont été rapportés3). Cependant, le protocole standard de Dresde nécessite une épaisseur cornéenne centrale ≥ 400 μm, et il est contre-indiqué pour les cornées très fines < 200 μm3). Des modifications du protocole pourraient élargir les indications, mais pour l’instant cela reste limité.
ZNF469 et PRDM5 codent tous deux des facteurs de transcription1). ZNF469 possède trois domaines en doigt de zinc de type C2H2 en C-terminal1). Il régule l’expression des gènes de la matrice extracellulaire (CLU, GPC6, PCOLCE2, THBS1)1). PRDM5 possède un domaine PR SET et régule directement la transcription des gènes du collagène via la liaison à l’ARN polymérase II4). Il est également impliqué dans la régulation de la voie de signalisation Wnt.
Ces mutations génétiques perturbent le dépôt de collagène et l’assemblage des fibrilles dans le stroma cornéen1). La mutation ZNF469 entraîne une diminution de l’expression du collagène de type I (COL-I) et des modifications structurelles 2). L’immunofluorescence du tissu cornéen après PKP a montré une diminution du COL-I et une augmentation du collagène de type III 2). La coloration de Masson a également révélé une nette réduction de la quantité de fibres de collagène 2).
La microscopie confocale révèle un réseau de structures linéaires hautement réfléchissantes dans le stroma cornéen antérieur 2). Il n’y a pas d’infiltration de cellules inflammatoires 2). Cela suggère que les lésions cornéennes de la BCS résultent d’une anomalie primaire de la structure du collagène.
Dans le tissu osseux, PRDM5 se lie également à l’ADN exonique du gène du collagène de type I 4). La biopsie osseuse a montré un amincissement de l’os cortical et une diminution marquée de la porosité corticale (1,3 % ; valeur normale 6,3 ± 0,6 %) 4). Un panel de 28 gènes de fragilité osseuse n’a détecté aucune autre mutation pathogène, suggérant que la mutation ZNF469 elle-même pourrait être la cause de la fragilité osseuse 4).
QPourquoi les tissus autres que la cornée sont-ils affectés dans la BCS ?
A
ZNF469 et PRDM5 sont impliqués dans la régulation transcriptionnelle de la matrice extracellulaire dans tout le tissu conjonctif 1). Les anomalies structurelles des fibres de collagène ne se limitent pas à la cornée ; elles affectent le tissu conjonctif dans tout le corps, y compris la sclère (décoloration bleue), les articulations (hypermobilité), la peau (hyperextensibilité), les os (ostéopénie) et le tympan (hypercompliance) 4). Des rapports d’ostéopénie et de fractures suggèrent que la BCS pourrait avoir un phénotype de fragilité osseuse 4).
Les progrès de la thérapie génique offrent de nouvelles possibilités pour la BCS. L’édition génomique par CRISPR et l’interférence ARN (ARNi) sont considérées comme des traitements prometteurs pour stabiliser ou améliorer l’amincissement cornéen 3).
Les implants cornéens bio-ingénierés (BPCDX) sont des hydrogels acellulaires transparents fabriqués à partir de collagène porcin médical 3). Il s’agit d’une procédure mini-invasive où l’implant est inséré dans une poche intrastromale de 2 à 3 mm créée par laser femtoseconde3). Une observation de 24 mois a rapporté une augmentation de l’épaisseur cornéenne, un aplatissement de 18 D de la kératométrie maximale et une amélioration de l’acuité visuelle corrigée3). Des résultats stables ont été obtenus sans rejet 3).
L’injection intrastromale de cellules souches mésenchymateuses est également étudiée 3). Les cellules souches dérivées du tissu adipeux et de la moelle osseuse sont injectées dans une incision lamellaire créée par laser femtoseconde, ce qui pourrait permettre de retarder la PKP3). Actuellement, cette technique est limitée aux patients atteints de kératocône, mais son application aux patients BCS est attendue 3).
Sanri A, Demir S, Gürkan H. Homozygous Val6Gly Variation in PRDM5 Gene Causing Brittle Cornea Syndrome: A New Turkish Case. Mol Syndromol. 2023;14(2):129-135. doi:10.1159/000524832. PMID:37064337; PMCID:PMC10091010.
Geng X, Zhu L, Li J, Li Z. Brittle cornea syndrome: A novel mutation. Heliyon. 2024;10:e32506. doi:10.1016/j.heliyon.2024.e32506.
Zeppieri M, Gentile M, Acquaviva A, Scollo D, D’Esposito F, Gagliano G, et al. Brittle Cornea Syndrome: Molecular Diagnosis and Management. Diagnostics (Basel, Switzerland). 2025;15(13). doi:10.3390/diagnostics15131596. PMID:40647596; PMCID:PMC12249002.
Cundy T, Vincent A, Robertson S. Does brittle cornea syndrome have a bone fragility phenotype? Bone Rep. 2021;15:101124. doi:10.1016/j.bonr.2021.101124. PMID:34522702; PMCID:PMC8426531.
Mandlik K, Betdur RA, Rashmita R, Narayana S. Brittle cornea syndrome: A tale of three brothers. Indian journal of ophthalmology. 2022;70(7):2594-2597. doi:10.4103/ijo.IJO_2894_21. PMID:35791165; PMCID:PMC9426125.
Copiez le texte de l'article et collez-le dans l'assistant IA de votre choix.
Article copié dans le presse-papiers
Ouvrez un assistant IA ci-dessous et collez le texte copié dans la conversation.