La sindrome della cornea fragile (Brittle Cornea Syndrome: BCS; OMIM 229200, 614170) è una rara malattia del tessuto connettivo a trasmissione autosomica recessiva caratterizzata da assottigliamento corneale progressivo e sclera blu. È stata riportata per la prima volta da Stein et al. nel 1968 2). La prevalenza è stimata in meno di 1 persona per milione 1). Al 2021 sono stati riportati complessivamente 86 casi, molti dei quali con storia familiare di consanguineità 1).
La BCS è classificata in 2 tipi in base al gene causativo 1). Entrambi i tipi codificano per fattori di trascrizione coinvolti nell’omeostasi della matrice extracellulare 1)2). I difetti nella deposizione del collagene e nell’assemblaggio delle fibre causano debolezza strutturale dello stroma corneale1).
BCS tipo 1 (mutazione ZNF469)
Gene causativo: ZNF469 (16q24), un gene a singolo esone che codifica per 3.953 amminoacidi 1)
Numero di pazienti riportati: 53 (24 mutazioni identificate) 1)
Principali tipi di mutazione: Frequenti mutazioni frameshift omozigoti o non senso 1)
Nota speciale: Le mutazioni eterozigoti sono state associate al cheratocono2)
BCS tipo 2 (mutazione PRDM5)
Gene causale: PRDM5 (4q27), un gene di 16 esoni che codifica per 630 amminoacidi1)
Numero di pazienti riportati: 33 (identificate 14 mutazioni)1)
Principali tipi di mutazione: Tutte riportate come mutazioni omozigoti1)
Nota speciale: Coinvolto anche nello sviluppo dei microvasi retinici e della membrana di Bruch1)
QIn cosa differisce la BCS dalla sindrome di Ehlers-Danlos?
A
La BCS era precedentemente considerata parte della sindrome di Ehlers-Danlos cifoscoliotica (EDS tipo VI). Tuttavia, le analisi di genetica molecolare hanno confermato che si tratta di una malattia distinta1). L’EDS VI è causato da un deficit di lisil idrossilasi dovuto a mutazioni del gene PLOD1. Può essere differenziato da un rapporto desossipiridinolina/piridinolina urinario elevato, mentre nella BCS questo rapporto è normale1). L’EDS VI ha una prognosi infausta per la vita a causa di rotture arteriose, mentre l’aspettativa di vita nella BCS è considerata normale.
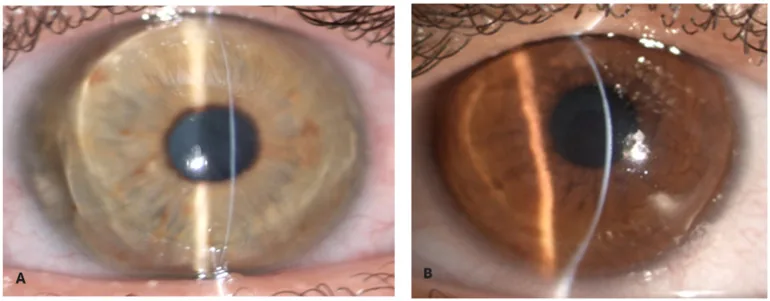
Zeppieri M, Nabil R, Ruzza A, et al. Brittle Cornea Syndrome: Molecular Diagnosis and Management. Diagnostics (Basel). 2025 Jun 24;15(13):1596. Figure 1. PMCID: PMC12249002. License: CC BY.
Con una stretta fenditura luminosa della lampada a fessura, lo stroma corneale appare estremamente sottile e rigonfio in avanti. L’estremo assottigliamento e la protrusione conica danno l’aspetto di una cornea fragile.
Il sintomo principale è una riduzione dell’acuità visiva dovuta a miopia progressiva o astigmatismo irregolare3). In caso di perforazione corneale, si verificano dolore improvviso e perdita della vista5). Può anche essere notata una perdita dell’udito a causa di sordità1).
L’assottigliamento dell’intera cornea (da limbo a limbo) è la caratteristica più tipica. Lo spessore corneale centrale (CCT) è spesso inferiore a 400 μm1). In un report su tre fratelli, il CCT era compreso tra 243 e 304 μm5). In fratelli albanesi, era di 189 μm e 157 μm3). In due fratelli neozelandesi, è stato confermato un assottigliamento estremo rispettivamente di 167 μm e 149 μm4).
La perforazione corneale si verifica in media a 4,3 anni (range 1,5–19 anni) 1). In oltre due terzi dei casi riportati si osserva perforazione del bulbo oculare 1). In più della metà dei casi si verifica una perdita permanente della vista 1). La sclera blu è il reperto oculare più frequente, osservato in 72 dei 78 casi 1).
L’ipermobilità articolare è il reperto extraoculare più frequente 1). Sono state riportate anche displasia congenita dell’anca, scoliosi e piedi piatti 1). La perdita dell’udito tende a colpire maggiormente le alte frequenze 1). È caratteristica un’ipercompiacenza della membrana timpanica 1). La pelle è morbida, iperestensibile e associata a facilità di ecchimosi 1).
Le segnalazioni di fragilità ossea sono aumentate anche negli ultimi anni. Due fratelli con mutazioni eterozigoti composte di ZNF469 hanno presentato più di 10 fratture e osteopenia 4). È stato suggerito che circa il 16% dei pazienti con BCS potrebbe presentare fragilità ossea 4). La biopsia ossea ha mostrato assottigliamento della corticale e una marcata riduzione della porosità corticale 4).
La BCS è una malattia autosomica recessiva causata da mutazioni bialleliche di due geni1).
ZNF469 è coinvolto nel normale sviluppo della camera anteriore e della cornea1). Studi di associazione genome-wide hanno mostrato un’associazione con lo spessore corneale centrale4). PRDM5 regola direttamente la trascrizione dei geni del collagene fibrillare4). Mutazioni con perdita di funzione (frameshift, nonsenso) sono frequenti in entrambi i geni1).
Le mutazioni eterozigoti di ZNF469 sono associate a una riduzione dello spessore corneale centrale2). Tuttavia, non sempre portano a un assottigliamento corneale. In un uomo cinese di 64 anni con mutazione eterozigote di ZNF469, lo spessore corneale era normale (circa 550 μm), ma si sono verificati opacità corneale e difetto epiteliale2).
La consanguineità è un importante fattore di rischio, aumentando la probabilità di mutazioni omozigoti1). In tre casi turchi, la mutazione PRDM5 c.17T>G, p.(Val6Gly) era comune, suggerendo un possibile effetto fondatore1).
La diagnosi clinica si basa su un assottigliamento corneale globale (spessore corneale centrale <400 μm), sclere blu e sintomi sistemici come ipermobilità articolare e perdita dell’udito3). Sono indispensabili la pachimetria corneale e l’analisi morfologica della cornea con Pentacam3). L’OCT del segmento anteriore è utile per una valutazione dettagliata della struttura corneale2)3).
La diagnosi definitiva richiede test genetici per ZNF469 e PRDM53). Il sequenziamento dell’intero esoma (WES) è efficace per identificare varianti patogene3). Fornisce anche informazioni utili per la consulenza genetica e la pianificazione familiare3). I portatori possono sviluppare miopia o lieve assottigliamento corneale2).
Dissezione aortica, alta statura, lussazione del cristallino
Nel BCS, la distinzione dall’EDS cifoscoliotico (kEDS-PLOD1) è particolarmente importante. Nel kEDS-PLOD1, se si verifica una rottura del bulbo oculare, la sclera è più soggetta a rottura rispetto alla cornea1). Nel kEDS-PLOD1, la scoliosi, l’ipotonia muscolare e le complicanze vascolari sono più marcate1).
QPerché è importante la diagnosi precoce del BCS?
A
L’età media di insorgenza della perforazione corneale è di 4,3 anni e la riparazione chirurgica dopo la perforazione è estremamente difficile5). In più della metà dei casi riportati si verifica una perdita permanente della vista1). Una diagnosi precoce consente l’uso di occhiali protettivi e l’educazione alla prevenzione dei traumi1). Lo screening dei fratelli è importante anche per la diagnosi precoce5).
L’aspetto più importante della gestione del BCS è la prevenzione della perforazione corneale1). Si raccomanda l’uso costante di occhiali protettivi in policarbonato5). Si considera anche la somministrazione profilattica di farmaci ipotensivi oculari. Le lenti a contatto sono limitate a causa dell’assottigliamento corneale e del rischio di trauma1).
Indicazione: Eseguita per grave assottigliamento corneale o per recupero visivo dopo perforazione3)
Tecnica chirurgica: utilizzo di un trefino da 8,0 mm, 16 suture interrotte in nylon 10-03)
Risultati: in fratelli albanesi, la migliore acuità visiva corretta (BCVA) è migliorata da 20/200 a 20/30 e 20/253)
Decorso: nessuna complicanza per 7 anni3)
Cheratoplastica lamellare profonda (DALK)
Vantaggi: preserva l’endotelio, riducendo il rischio di rigetto3)
Limitazioni: rischio di perforazione intraoperatoria a causa della fragilità dello stroma3)
Caso riportato: in un caso si è verificata una perforazione endoteliale centrale intraoperatoria, convertita in PKP3)
Criteri di selezione: spessore stromale sufficiente e integrità della membrana di Descemet necessari3)
Nella riparazione delle perforazioni corneali, l’estrema fragilità tissutale è un problema. In un report su tre fratelli, l’uso di punti di sutura lunghi, colla cianoacrilica e lenti a contatto medicate è stato efficace5). La creazione attenta di un tunnel corneosclerale può ridurre le complicanze5). In un paziente BCS di 64 anni è stata eseguita PKP e l’acuità visiva corretta alla dimissione era 0,22).
QIl cross-linking del collagene corneale è efficace?
A
Due pazienti pediatrici BCS con spessore corneale centrale < 280 μm sono stati sottoposti a CXL transepiteliale (con aggiustamento della dose UV in base allo spessore corneale)3). Sono stati riportati miglioramento dell’acuità visiva e mantenimento della densità delle cellule endoteliali3). Tuttavia, il protocollo standard di Dresda richiede uno spessore corneale centrale ≥ 400 μm ed è controindicato per cornee molto sottili < 200 μm3). Modifiche del protocollo potrebbero ampliare le indicazioni, ma al momento è limitato.
6. Fisiopatologia e meccanismi dettagliati della patogenesi
ZNF469 e PRDM5 codificano entrambi fattori di trascrizione1). ZNF469 ha tre domini a dito di zinco di tipo C2H2 all’estremità C-terminale1). Regola l’espressione dei geni della matrice extracellulare (CLU, GPC6, PCOLCE2, THBS1)1). PRDM5 possiede un dominio PR SET e regola direttamente la trascrizione dei geni del collagene tramite il legame con l’RNA polimerasi II4). È anche coinvolto nella regolazione della via di segnalazione Wnt.
Queste mutazioni genetiche alterano la deposizione di collagene e l’assemblaggio delle fibrille nello stroma corneale1). La mutazione ZNF469 causa una ridotta espressione del collagene di tipo I (COL-I) e alterazioni strutturali 2). L’immunofluorescenza del tessuto corneale dopo PKP ha mostrato una diminuzione del COL-I e un aumento del collagene di tipo III 2). Anche la colorazione di Masson ha evidenziato una chiara riduzione della quantità di fibre di collagene 2).
La microscopia confocale rivela una rete di strutture lineari altamente riflettenti nello stroma corneale anteriore 2). Non vi è infiltrazione di cellule infiammatorie 2). Ciò suggerisce che le lesioni corneali nella BCS siano dovute a un’anomalia primaria della struttura del collagene.
Nel tessuto osseo, anche PRDM5 si lega al DNA esonico del gene del collagene di tipo I 4). La biopsia ossea ha mostrato un assottigliamento dell’osso corticale e una marcata riduzione della porosità corticale (1,3%; valore normale 6,3±0,6%) 4). Un pannello di 28 geni di fragilità ossea non ha rilevato altre mutazioni patogene, suggerendo che la mutazione ZNF469 stessa potrebbe essere la causa della fragilità ossea 4).
QPerché nella BCS sono colpiti anche tessuti diversi dalla cornea?
A
ZNF469 e PRDM5 sono coinvolti nella regolazione trascrizionale della matrice extracellulare in tutto il tessuto connettivo 1). Le anomalie strutturali delle fibre di collagene non sono limitate alla cornea; interessano il tessuto connettivo in tutto il corpo, inclusi sclera (colorazione blu), articolazioni (ipermobilità), pelle (iperestensibilità), ossa (osteopenia) e timpano (ipercompliance) 4). Segnalazioni di osteopenia e fratture suggeriscono che la BCS potrebbe avere un fenotipo di fragilità ossea 4).
I progressi nella terapia genica stanno aprendo nuove possibilità per la BCS. L’editing genomico CRISPR e l’interferenza a RNA (RNAi) sono considerati trattamenti promettenti per stabilizzare o migliorare l’assottigliamento corneale 3).
Gli impianti corneali bioingegnerizzati (BPCDX) sono idrogel trasparenti acellulari realizzati con collagene suino di grado medico 3). Si tratta di una procedura mini-invasiva in cui l’impianto viene inserito in una tasca intrastromale di 2-3 mm creata con laser a femtosecondi3). Un’osservazione di 24 mesi ha riportato un aumento dello spessore corneale, un appiattimento di 18 D della cheratometria massima e un miglioramento dell’acuità visiva corretta3). Sono stati ottenuti risultati stabili senza rigetto 3).
È in studio anche l’iniezione intrastromale di cellule staminali mesenchimali 3). Le cellule staminali derivate dal tessuto adiposo e dal midollo osseo vengono iniettate in un’incisione lamellare creata con laser a femtosecondi, che potrebbe consentire di ritardare la PKP3). Attualmente questa tecnica è limitata ai pazienti con cheratocono, ma si prevede la sua applicazione nei pazienti con BCS 3).
Sanri A, Demir S, Gürkan H. Homozygous Val6Gly Variation in PRDM5 Gene Causing Brittle Cornea Syndrome: A New Turkish Case. Mol Syndromol. 2023;14(2):129-135. doi:10.1159/000524832. PMID:37064337; PMCID:PMC10091010.
Geng X, Zhu L, Li J, Li Z. Brittle cornea syndrome: A novel mutation. Heliyon. 2024;10:e32506. doi:10.1016/j.heliyon.2024.e32506.
Zeppieri M, Gentile M, Acquaviva A, Scollo D, D’Esposito F, Gagliano G, et al. Brittle Cornea Syndrome: Molecular Diagnosis and Management. Diagnostics (Basel, Switzerland). 2025;15(13). doi:10.3390/diagnostics15131596. PMID:40647596; PMCID:PMC12249002.
Cundy T, Vincent A, Robertson S. Does brittle cornea syndrome have a bone fragility phenotype? Bone Rep. 2021;15:101124. doi:10.1016/j.bonr.2021.101124. PMID:34522702; PMCID:PMC8426531.
Mandlik K, Betdur RA, Rashmita R, Narayana S. Brittle cornea syndrome: A tale of three brothers. Indian journal of ophthalmology. 2022;70(7):2594-2597. doi:10.4103/ijo.IJO_2894_21. PMID:35791165; PMCID:PMC9426125.
Copia il testo dell'articolo e incollalo nell'assistente IA che preferisci.
Articolo copiato negli appunti
Apri un assistente IA qui sotto e incolla il testo copiato nella chat.