กลุ่มอาการกระจกตา เปราะ (BCS ) เป็นโรคเนื้อเยื่อเกี่ยวพันที่ถ่ายทอดทางพันธุกรรมแบบออโตโซมด้อย เกิดจากการกลายพันธุ์ของยีน ZNF 469 หรือ PRDM5 ทั้งสองอัลลีล โดยมีความชุกน้อยกว่า 1 ต่อ 1,000,000 คน 1) .
มีลักษณะเด่นคือกระจกตา บางลงทั่วทั้งแผ่น (ความหนากระจกตา ส่วนกลางน้อยกว่า 400 ไมโครเมตร) ทำให้เกิดการทะลุของกระจกตา ได้จากการบาดเจ็บเล็กน้อยหรือแตกเอง 1) .
เป็นโรคที่มีหลายอวัยวะเกี่ยวข้อง ร่วมกับอาการทั่วร่างกาย เช่น ตาขาว สีน้ำเงิน ข้อต่อยืดหยุ่นเกินปกติ การได้ยินลดลง และผิวหนังยืดเกิน 1) .
อายุเฉลี่ยที่เกิดการทะลุของกระจกตา ประมาณ 4.3 ปี และพบการทะลุในมากกว่าสองในสามของรายงานผู้ป่วย 1) .
เสาหลักของการรักษาคือการวินิจฉัยตั้งแต่เนิ่นๆ และการใช้แว่นตาป้องกันเพื่อป้องกันการทะลุ มีรายงานการฟื้นฟูการมองเห็น ด้วยการปลูกถ่ายกระจกตา แบบเต็มความหนา 3) .
กลุ่มอาการกระจกตา เปราะ (Brittle Cornea Syndrome: BCS ; OMIM 229200, 614170) เป็นโรคเนื้อเยื่อเกี่ยวพันที่ถ่ายทอดทางพันธุกรรมแบบออโตโซมด้อยที่พบได้ยาก มีลักษณะเด่นคือกระจกตา บางลงเรื่อยๆ และตาขาว สีน้ำเงิน รายงานครั้งแรกโดย Stein และคณะในปี 1968 2) ความชุกประมาณน้อยกว่า 1 ต่อ 1,000,000 คน 1) จนถึงปี 2021 มีรายงานผู้ป่วยสะสม 86 ราย โดยหลายรายมีประวัติครอบครัวแต่งงานในเครือญาติ 1) .
BCS แบ่งออกเป็น 2 ชนิดตามยีนที่เป็นสาเหตุ 1) ทั้งสองชนิดเข้ารหัสปัจจัยการถอดรหัสที่เกี่ยวข้องกับการรักษาสมดุลของเมทริกซ์นอกเซลล์ 1) 2) ความบกพร่องในการสะสมคอลลาเจนและการรวมตัวของเส้นใยทำให้เกิดความอ่อนแอทางโครงสร้างของสโตรมาของกระจกตา 1) .
BCS ชนิดที่ 1 (การกลายพันธุ์ของ ZNF469)
ยีนที่เป็นสาเหตุ : ยีน ZNF 469 (16q24) ซึ่งเป็นยีนเอกซอนเดี่ยว เข้ารหัสกรดอะมิโน 3,953 ตัว 1)
จำนวนผู้ป่วยที่รายงาน : 53 ราย (ระบุการกลายพันธุ์ได้ 24 ชนิด) 1)
การกลายพันธุ์หลัก : ส่วนใหญ่เป็นการกลายพันธุ์แบบเฟรมชิฟต์แบบโฮโมไซกัสหรือการกลายพันธุ์แบบนอนเซนส์ 1)
หมายเหตุพิเศษ : มีรายงานว่าการกลายพันธุ์แบบเฮเทอโรไซกัสมีความสัมพันธ์กับโรคกระจกตา รูปกรวย 2)
BCS ชนิด 2 (การกลายพันธุ์ PRDM5)
ยีนก่อโรค : PRDM5 (4q27) เป็นยีน 16 เอ็กซอนที่เข้ารหัสกรดอะมิโน 630 ตัว 1)
จำนวนผู้ป่วยที่รายงาน : 33 ราย (ระบุการกลายพันธุ์ 14 ชนิด) 1)
ชนิดการกลายพันธุ์หลัก : ทั้งหมดรายงานว่าเป็นการกลายพันธุ์แบบโฮโมไซกัส 1)
หมายเหตุพิเศษ : ยังเกี่ยวข้องกับการพัฒนาของหลอดเลือดขนาดเล็กในจอตาและเยื่อบรูช 1)
Q
BCS แตกต่างจากกลุ่มอาการเอห์เลอร์ส-ดานลอสอย่างไร?
A
ก่อนหน้านี้ BCS ถูกถือว่าเป็นส่วนหนึ่งของกลุ่มอาการเอห์เลอร์ส-ดานลอส ชนิดคิฟอสโคลิโอติก (EDS VI) อย่างไรก็ตาม การวิเคราะห์ทางอณูพันธุศาสตร์ยืนยันว่าเป็นโรคที่แตกต่างกัน 1) EDS VI เกิดจากการขาดเอนไซม์ไลซิลไฮดรอกซีเลสเนื่องจากการกลายพันธุ์ของยีน PLOD1 สามารถแยกความแตกต่างได้โดยอัตราส่วนดีออกซีไพริดิโนลีน/ไพริดิโนลีนในปัสสาวะที่สูงขึ้น ในขณะที่ BCS อัตราส่วนนี้ปกติ 1) EDS VI มีการพยากรณ์โรคที่ไม่ดีเนื่องจากการแตกของหลอดเลือดแดง ในขณะที่อายุขัยเฉลี่ยใน BCS ปกติ
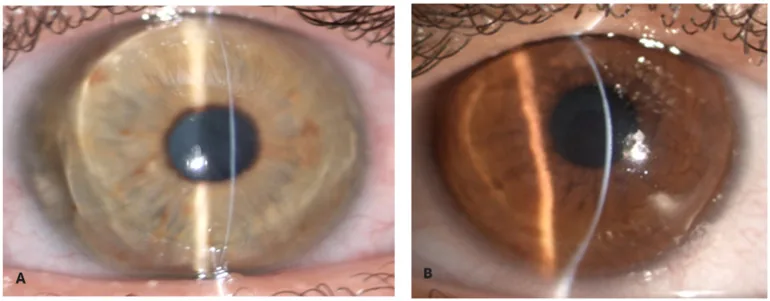
Zeppieri M, Nabil R, Ruzza A, et al. Brittle Cornea Syndrome: Molecular Diagnosis and Management. Diagnostics (Basel). 2025 Jun 24;15(13):1596. Figure 1. PM
CI D: PMC12249002. License: CC BY.
ด้วยแสงสลิตแคบจากโคมไฟกรีด สโตรมาของกระจกตา ปรากฏบางมากและโป่งไปข้างหน้า การบางลงอย่างรุนแรงและการยื่นเป็นรูปกรวยแสดงให้เห็นลักษณะของกระจกตา ที่เปราะ
ข้อร้องเรียนหลักคือการมองเห็น ลดลงเนื่องจากสายตาสั้น ที่ลุกลามหรือสายตาเอียง ไม่สม่ำเสมอ 3) เมื่อเกิดการทะลุของกระจกตา จะมีอาการปวดอย่างฉับพลันและสูญเสียการมองเห็น 5) ผู้ป่วยอาจสังเกตเห็นความบกพร่องทางการได้ยินเนื่องจากหูหนวก 1)
ลักษณะเด่นที่สุดคือกระจกตา บางทั่วทั้งแผ่น (จากลิมบัส ถึงลิมบัส ) ความหนากระจกตา ส่วนกลาง (CCT) มักน้อยกว่า 400 ไมโครเมตร 1) ในรายงานพี่น้องสามคน CCT อยู่ระหว่าง 243 ถึง 304 ไมโครเมตร 5) ในพี่น้องชาวแอลเบเนีย วัดได้ 189 ไมโครเมตรและ 157 ไมโครเมตร 3) ในพี่น้องชาวนิวซีแลนด์สองคน ยืนยันการบางลงอย่างรุนแรงที่ 167 ไมโครเมตรและ 149 ไมโครเมตรตามลำดับ 4)
การทะลุของกระจกตา เกิดขึ้นที่อายุเฉลี่ย 4.3 ปี (ช่วง 1.5–19 ปี) 1) พบการทะลุของลูกตาในมากกว่าสองในสามของรายงานผู้ป่วย 1) การสูญเสียการมองเห็น ถาวรเกิดขึ้นในมากกว่าครึ่งหนึ่งของผู้ป่วย 1) ตาขาว สีฟ้าเป็นอาการทางตาที่พบบ่อยที่สุด พบใน 72 จาก 78 ราย 1)
ระบบ อาการหลัก ความถี่ ตา กระจกตา บาง/ทะลุ ตาขาว สีฟ้า>90% 1) ข้อต่อ ข้อต่อเล็กเคลื่อนไหวเกิน 64/78 ราย 1) การได้ยิน การสูญเสียการได้ยินแบบประสาทรับเสียง แบบนำเสียง และแบบผสม 32/78 ราย 1)
ข้อต่อเคลื่อนไหวเกินเป็นอาการนอกตาที่พบบ่อยที่สุด 1) มีรายงานภาวะสะโพกเคลื่อนแต่กำเนิด กระดูกสันหลังคด และเท้าแบนด้วย 1) การสูญเสียการได้ยินมักรุนแรงกว่าที่ความถี่สูง 1) เยื่อแก้วหูมีความยืดหยุ่นเกินเป็นลักษณะเฉพาะ 1) ผิวนุ่มและยืดเกินปกติร่วมกับมีแนวโน้มเลือดออกง่าย 1)
รายงานความเปราะบางของกระดูกก็เพิ่มขึ้นในช่วงไม่กี่ปีที่ผ่านมา พี่น้องสองคนที่มีการกลายพันธุ์แบบ compound heterozygous ในยีน ZNF 469 มีประวัติกระดูกหักมากกว่า 10 ครั้งและภาวะกระดูกบาง 4) มีการเสนอว่าผู้ป่วย BCS ประมาณ 16% อาจมีความเปราะบางของกระดูก 4) การตัดชิ้นเนื้อกระดูกพบว่ากระดูกคอร์เทกซ์บางลงและความพรุนของกระดูกคอร์เทกซ์ลดลงอย่างชัดเจน 4)
BCS เป็นโรคถ่ายทอดทางพันธุกรรมแบบออโตโซมัลรีเซสซีฟที่เกิดจากการกลายพันธุ์ของยีนสองยีนในทั้งสองอัลลีล 1) .
ZNF 469 เกี่ยวข้องกับการพัฒนาปกติของช่องหน้าม่านตา และกระจกตา 1) การศึกษา association ทั่วทั้งจีโนมแสดงความสัมพันธ์กับความหนากระจกตา ส่วนกลาง 4) PRDM5 ควบคุมการถอดรหัสของยีนคอลลาเจนชนิดเส้นใยโดยตรง 4) การกลายพันธุ์แบบสูญเสียหน้าที่ (frameshift และ nonsense) พบได้บ่อยในยีนทั้งสอง 1) .
การกลายพันธุ์แบบเฮเทอโรไซกัสใน ZNF 469 สัมพันธ์กับความหนากระจกตา ส่วนกลางที่ลดลง 2) อย่างไรก็ตาม ไม่ได้ทำให้กระจกตา บางลงเสมอไป ในชายชาวจีนอายุ 64 ปี การกลายพันธุ์ ZNF 469 แบบเฮเทอโรไซกัสทำให้ความหนากระจกตา ปกติ (ประมาณ 550 μm) แต่เขามีกระจกตา ขุ่นและข้อบกพร่องของเยื่อบุผิว 2) .
การแต่งงานในเครือญาติเป็นปัจจัยเสี่ยงสำคัญ เพิ่มโอกาสเกิดการกลายพันธุ์แบบโฮโมไซกัส 1) ในสามกรณีเชื้อสายตุรกี พบการกลายพันธุ์ PRDM5 c.17T>G, p.(Val6Gly) ร่วมกัน ซึ่งบ่งชี้ถึงการกลายพันธุ์ของผู้ก่อตั้ง 1) .
แนะนำให้สวมแว่นตาป้องกันโพลีคาร์บอเนตตลอดเวลา การหลีกเลี่ยงกีฬาที่มีการปะทะและการจำกัดการขยี้ตาเป็นสิ่งสำคัญ 1) การให้ความรู้เกี่ยวกับโรคแก่ผู้ปกครองและบุคลากรโรงเรียนก็จำเป็น 5) ควรตรวจตาเป็นประจำและประเมินระบบอื่นๆ รวมถึงการได้ยินและความหนาแน่นของกระดูกอย่างต่อเนื่อง
การวินิจฉัยทางคลินิกขึ้นอยู่กับกระจกตา บางทั่วทั้งแผ่น (ความหนากระจกตา ส่วนกลางน้อยกว่า 400 μm) และตาขาว สีน้ำเงิน ร่วมกับอาการทางระบบ เช่น ข้อต่อยืดหยุ่นเกินและการสูญเสียการได้ยิน 3) การวัดความหนากระจกตา และการวิเคราะห์รูปร่างกระจกตา ด้วย Pentacam เป็นสิ่งจำเป็น 3) OCT ส่วนหน้าตาเป็นประโยชน์ในการประเมินโครงสร้างกระจกตา อย่างละเอียด 2) 3) .
เพื่อการวินิจฉัยที่แน่นอน จำเป็นต้องตรวจทางพันธุกรรมของยีน ZNF 469 และ PRDM5 3) การหาลำดับเอ็กโซมทั้งหมด (WES) มีประสิทธิภาพในการระบุตัวแปรที่ก่อโรค 3) นอกจากนี้ยังให้ข้อมูลที่เป็นประโยชน์สำหรับการให้คำปรึกษาทางพันธุกรรม และการวางแผนครอบครัว 3) ผู้ที่มียีนกลายพันธุ์อาจเกิดสายตาสั้น หรือกระจกตา บางเล็กน้อย 2) .
โรคที่ต้องแยก ความแตกต่างจาก BCS EDS ชนิดกระดูกสันหลังคดและหลังโกง ความเสี่ยงต่อการแตกของหลอดเลือดแดง, อัตราส่วนไลซิลไพริดิโนลีนในปัสสาวะสูงขึ้น1) โรคกระดูกเปราะ กระดูกหักซ้ำเป็นอาการหลัก, ฟันเจริญผิดปกติ4) กลุ่มอาการมาร์แฟน การผ่าหลอดเลือดเอออร์ตา, รูปร่างสูง, เลนส์ตาเคลื่อน
ใน BCS การแยกความแตกต่างจาก EDS ชนิดกระดูกสันหลังคดและหลังโกง (kEDS-PLOD1) มีความสำคัญเป็นพิเศษ ใน kEDS-PLOD1 หากเกิดการแตกของลูกตา ตาขาว จะแตกง่ายกว่ากระจกตา 1) ใน kEDS-PLOD1 กระดูกสันหลังคด กล้ามเนื้ออ่อนแรง และภาวะแทรกซ้อนทางหลอดเลือดจะเด่นชัดกว่า1)
Q
เหตุใดการวินิจฉัย BCS ตั้งแต่เนิ่นๆ จึงสำคัญ?
A
อายุเฉลี่ยที่เกิดการทะลุของกระจกตา คือ 4.3 ปี และการซ่อมแซมหลังการทะลุทำได้ยากมาก5) มากกว่าครึ่งหนึ่งของรายงานผู้ป่วยสูญเสียการมองเห็น ถาวร1) การวินิจฉัยตั้งแต่เนิ่นๆ ช่วยให้สามารถสวมแว่นตาป้องกันและให้ความรู้เกี่ยวกับการป้องกันการบาดเจ็บ1) การตรวจคัดกรองพี่น้องก็มีความสำคัญต่อการตรวจพบตั้งแต่เนิ่นๆ เช่นกัน5)
สิ่งที่สำคัญที่สุดในการดูแล BCS คือการป้องกันการทะลุของกระจกตา 1) แนะนำให้สวมแว่นตาป้องกันที่ทำจากโพลีคาร์บอเนตตลอดเวลา5) อาจพิจารณาใช้ยาหยอดตาลดความดันลูกตา เพื่อป้องกัน คอนแทคเลนส์มีข้อจำกัดเนื่องจากกระจกตา บางและเสี่ยงต่อการบาดเจ็บ1)
การปลูกถ่ายกระจกตาแบบทะลุทะลวง (PKP)
ข้อบ่งชี้ : ทำเพื่อฟื้นฟูการมองเห็น ในกรณีกระจกตา บางมากหรือหลังการทะลุ3)
เทคนิคการผ่าตัด : ใช้ trephine ขนาด 8.0 มม. เย็บไนลอน 10-0 แบบ interrupted 16 เข็ม3)
ผลลัพธ์ : การมองเห็น ที่ดีที่สุดที่แก้ไขแล้ว (BCVA) ดีขึ้นจาก 20/200 เป็น 20/30 และ 20/25 ในพี่น้องชาวแอลเบเนียสองคน3)
การดำเนินโรค : 7 ปีโดยไม่มีภาวะแทรกซ้อน3)
การปลูกถ่ายกระจกตาชั้นลึก (DALK)
ข้อดี : การรักษาชั้น内皮ไว้ช่วยลดความเสี่ยงของการปฏิเสธ3)
ข้อจำกัด : ความเปราะบางของชั้นสโตรมาทำให้เสี่ยงต่อการทะลุระหว่างผ่าตัด3)
รายงานผู้ป่วย : ในหนึ่งรายเกิดการทะลุของชั้น内皮ส่วนกลางระหว่างผ่าตัดและเปลี่ยนเป็น PKP 3)
เกณฑ์การเลือก : ต้องมีความหนาของสโตรมาเพียงพอและความสมบูรณ์ของเยื่อหุ้มเดสเซเมท3)
ในการซ่อมแซมกระจกตา ทะลุ ความเปราะบางของเนื้อเยื่ออย่างรุนแรงเป็นปัญหา ในรายงานพี่น้องสามคน การใช้ไหมเย็บแบบกัดยาว กาวไซยาโนอะคริเลต และคอนแทคเลนส์ปิดแผลมีประสิทธิภาพ5) การสร้างอุโมงค์กระจกตา -ตาขาว อย่างระมัดระวังสามารถลดภาวะแทรกซ้อนได้5) ในผู้ป่วย BCS อายุ 64 ปี ได้ทำ PKP และค่าสายตาที่แก้ไขแล้วเมื่อจำหน่ายคือ 0.22)
Q
การเชื่อมขวางคอลลาเจนกระจกตา (CXL) มีประสิทธิภาพหรือไม่?
A
ทำ CXL ผ่านเยื่อบุผิว (ปรับปริมาณรังสี UV ตามความหนากระจกตา ) ในผู้ป่วยเด็ก BCS 2 รายที่มีความหนากระจกตา ส่วนกลางน้อยกว่า 280 μm3) มีรายงานการมองเห็น ดีขึ้นและรักษาความหนาแน่นของเซลล์ชั้น内皮3) อย่างไรก็ตาม โปรโตคอลเดรสเดนมาตรฐานต้องการความหนากระจกตา ส่วนกลางอย่างน้อย 400 μm และไม่เหมาะกับกระจกตา ที่บางมากน้อยกว่า 200 μm3) การปรับเปลี่ยนโปรโตคอลคาดว่าจะขยายขอบเขตการใช้งาน แต่ปัจจุบันยังจำกัด
ZNF 469 และ PRDM5 ต่างก็เข้ารหัสปัจจัยการถอดรหัส1) ZNF 469 มีโดเมนซิงก์ฟิงเกอร์ชนิด C2H2 สามโดเมนที่ปลาย C1) ควบคุมการแสดงออกของยีนเมทริกซ์นอกเซลล์ (CLU, GPC 6, PCOLCE2, THBS1)1) PRDM5 มีโดเมน PR SET และควบคุมการถอดรหัสยีนคอลลาเจนโดยตรงผ่านการจับกับ RNA polymerase II4) ยังเกี่ยวข้องกับการควบคุมเส้นทางสัญญาณ Wnt
การกลายพันธุ์ของยีนเหล่านี้ทำให้การสะสมคอลลาเจนและการประกอบเส้นใยในสโตรมาของกระจกตา บกพร่อง 1) การกลายพันธุ์ของ ZNF 469 ทำให้การแสดงออกของคอลลาเจนชนิดที่ 1 (COL-I) ลดลงและการเปลี่ยนแปลงโครงสร้าง 2) การย้อมอิมมูโนฟลูออเรสเซนซ์ของเนื้อเยื่อกระจกตา หลัง PKP แสดงให้เห็นการลดลงของ COL-I และการเพิ่มขึ้นของคอลลาเจนชนิดที่ 3 2) การย้อม Masson ยังแสดงให้เห็นการลดลงอย่างชัดเจนของปริมาณเส้นใยคอลลาเจน 2)
กล้องจุลทรรศน์คอนโฟคอล แสดงเนื้อเยื่อเส้นใยสะท้อนแสงสูงเป็นร่างแหในสโตรมาส่วนหน้าของกระจกตา 2) ไม่มีการแทรกซึมของเซลล์อักเสบ 2) ซึ่งบ่งชี้ว่ารอยโรคที่กระจกตา ใน BCS เกิดจากความผิดปกติปฐมภูมิของโครงสร้างคอลลาเจน
ในเนื้อเยื่อกระดูก PRDM5 ยังจับกับ DNA เอ็กซอนของยีนคอลลาเจนชนิดที่ 1 4) การตัดชิ้นเนื้อกระดูกพบว่ากระดูกคอร์ติคอลบางลงและความพรุนของกระดูกคอร์ติคอลลดลงอย่างมีนัยสำคัญ (1.3%; ปกติ 6.3±0.6%) 4) แผงความเปราะบางของกระดูก 28 ยีนไม่พบการกลายพันธุ์ที่ก่อโรคอื่นๆ ซึ่งบ่งชี้ว่าการกลายพันธุ์ของ ZNF 469 เองอาจเป็นสาเหตุของความเปราะบางของกระดูก 4)
Q
ทำไมเนื้อเยื่ออื่นที่ไม่ใช่กระจกตาใน BCS จึงได้รับความเสียหาย?
A
ZNF 469 และ PRDM5 เกี่ยวข้องกับการควบคุมการถอดรหัสของเมทริกซ์นอกเซลล์ในเนื้อเยื่อเกี่ยวพันทั่วร่างกาย 1) ความผิดปกติของโครงสร้างเส้นใยคอลลาเจนไม่ได้จำกัดอยู่ที่กระจกตา แต่ส่งผลต่อเนื้อเยื่อเกี่ยวพันทั่วร่างกาย เช่น ตาขาว (สีน้ำเงิน), ข้อต่อ (เคลื่อนไหวมากเกิน), ผิวหนัง (ยืดเกิน), กระดูก (มวลกระดูกต่ำ), และเยื่อแก้วหู (ยืดหยุ่นเกิน) 4) มีรายงานภาวะมวลกระดูกต่ำและกระดูกหัก ซึ่งบ่งชี้ว่า BCS อาจมีฟีโนไทป์ของกระดูกเปราะบาง 4)
ความก้าวหน้าของการบำบัดด้วยยีน กำลังเปิดโอกาสใหม่สำหรับ BCS การแก้ไขจีโนมด้วย CRISPR และการแทรกแซง RNA (RNAi) คาดว่าจะเป็นการรักษาที่มีเป้าหมายเพื่อทำให้กระจกตา บางลงคงที่หรือดีขึ้น 3)
การปลูกถ่ายกระจกตา ที่สร้างขึ้นทางวิศวกรรมชีวภาพ (BPCDX) เป็นไฮโดรเจลใสไร้เซลล์ที่ทำจากคอลลาเจนหมูทางการแพทย์ 3) เป็นเทคนิคการรุกรานน้อยที่สุดโดยสอดใส่ในช่องในสโตรมาขนาด 2-3 มม. ที่สร้างด้วยเลเซอร์เฟมโตวินาที 3) การติดตามผล 24 เดือนพบว่าความหนาของกระจกตา เพิ่มขึ้น การแบนลง 18D ของค่าเคอราโตเมทรีสูงสุด และการมองเห็น ที่แก้ไขดีขึ้น 3) ผลลัพธ์ที่คงที่โดยไม่มีการปฏิเสธเกิดขึ้น 3)
การฉีดสเต็มเซลล์มีเซนไคม์เข้าไปในสโตรมาของกระจกตา กำลังอยู่ระหว่างการศึกษา 3) เทคนิคนี้เกี่ยวข้องกับการฉีดสเต็มเซลล์จากไขมันหรือไขกระดูกเข้าไปในรอยผ่าชั้นกระจกตา ด้วยเลเซอร์เฟมโตวินาที ซึ่งอาจชะลอความจำเป็นในการทำ PKP 3) ปัจจุบันการใช้จำกัดเฉพาะผู้ป่วยโรคกระจกตา โป่งพอง แต่คาดว่าจะนำไปใช้กับผู้ป่วย BCS 3)
Sanri A, Demir S, Gürkan H. Homozygous Val6Gly Variation in PRDM5 Gene Causing Brittle Cornea Syndrome: A New Turkish Case. Mol Syndromol. 2023;14(2):129-135. doi:10.1159/000524832. PMID:37064337; PMCI D:PMC10091010.
Geng X, Zhu L, Li J, Li Z. Brittle cornea syndrome: A novel mutation. Heliyon. 2024;10:e32506. doi:10.1016/j.heliyon.2024.e32506.
Zeppieri M, Gentile M, Acquaviva A, Scollo D, D’Esposito F, Gagliano G, et al. Brittle Cornea Syndrome: Molecular Diagnosis and Management. Diagnostics (Basel, Switzerland). 2025;15(13). doi:10.3390/diagnostics15131596. PMID:40647596; PMCI D:PMC12249002.
Cundy T, Vincent A, Robertson S. Does brittle cornea syndrome have a bone fragility phenotype? Bone Rep. 2021;15:101124. doi:10.1016/j.bonr.2021.101124. PMID:34522702; PMCI D:PMC8426531.
Mandlik K, Betdur RA, Rashmita R, Narayana S. Brittle cornea syndrome: A tale of three brothers. Indian journal of ophthalmology. 2022;70(7):2594-2597. doi:10.4103/ijo.IJO_2894_21. PMID:35791165; PMCI D:PMC9426125.