Das Brittle-Cornea-Syndrom (BCS; OMIM 229200, 614170) ist eine seltene autosomal-rezessive Bindegewebserkrankung, die durch eine fortschreitende Hornhautverdünnung und blaue Sklera gekennzeichnet ist. Es wurde erstmals 1968 von Stein et al. beschrieben 2). Die Prävalenz wird auf weniger als 1 Person pro Million geschätzt 1). Bis 2021 wurden insgesamt 86 Fälle berichtet, von denen viele eine familiäre Vorgeschichte von Konsanguinität aufweisen 1).
Das BCS wird je nach ursächlichem Gen in 2 Typen eingeteilt 1). Beide Typen kodieren für Transkriptionsfaktoren, die an der Homöostase der extrazellulären Matrix beteiligt sind 1)2). Störungen der Kollagenablagerung und Faseranordnung führen zu einer strukturellen Schwäche des Hornhautstromas 1).
BCS Typ 1 (ZNF469-Mutation)
Ursächliches Gen: ZNF469 (16q24), ein Einzelexon-Gen, das 3.953 Aminosäuren kodiert 1)
Hauptmutationstypen: Alle als homozygote Mutationen berichtet1)
Besonderer Hinweis: Auch an der Entwicklung von Netzhautmikrogefäßen und der Bruch-Membran beteiligt1)
QWie unterscheidet sich BCS vom Ehlers-Danlos-Syndrom?
A
BCS wurde früher als Teil des kyphoskoliotischen Ehlers-Danlos-Syndroms (EDS Typ VI) angesehen. Molekulargenetische Analysen haben jedoch bestätigt, dass es sich um eine eigenständige Erkrankung handelt1). EDS VI wird durch einen Lysylhydroxylase-Mangel aufgrund von PLOD1-Genmutationen verursacht. Es kann durch einen erhöhten Desoxypyridinolin/Pyridinolin-Quotienten im Urin unterschieden werden, während dieser Quotient bei BCS normal ist1). EDS VI hat aufgrund von Arterienrupturen eine schlechte Lebenserwartung, während die Lebenserwartung bei BCS als normal angesehen wird.
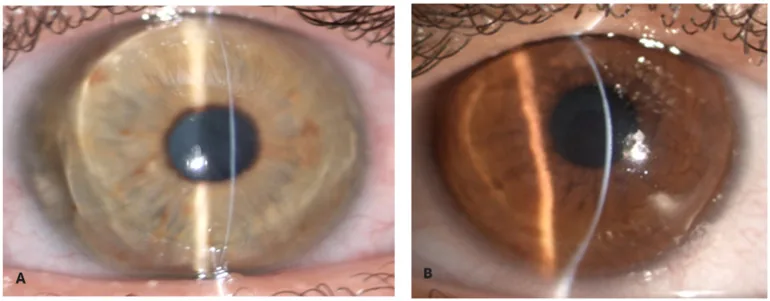
Zeppieri M, Nabil R, Ruzza A, et al. Brittle Cornea Syndrome: Molecular Diagnosis and Management. Diagnostics (Basel). 2025 Jun 24;15(13):1596. Figure 1. PMCID: PMC12249002. License: CC BY.
Mit einem schmalen Spaltlampenlicht erscheint das Hornhautstroma extrem dünn und nach vorne gewölbt. Die extreme Verdünnung und konische Vorwölbung zeigen das Erscheinungsbild einer brüchigen Hornhaut.
Die Hauptbeschwerde ist eine Verschlechterung der Sehschärfe aufgrund fortschreitender Kurzsichtigkeit oder unregelmäßigem Astigmatismus3). Bei einer Hornhautperforation treten plötzliche Schmerzen und Sehverlust auf5). Auch Hörverlust durch Taubheit kann bemerkt werden1).
Die Verdünnung der gesamten Hornhaut (von Limbus zu Limbus) ist am charakteristischsten. Die zentrale Hornhautdicke (CCT) beträgt oft weniger als 400 μm1). In einem Bericht über drei Brüder lag die CCT zwischen 243 und 304 μm5). Bei albanischen Geschwistern betrug sie 189 μm und 157 μm3). Bei zwei neuseeländischen Brüdern wurde eine extreme Verdünnung von 167 μm bzw. 149 μm bestätigt4).
Die Hornhautperforation tritt im Durchschnitt im Alter von 4,3 Jahren auf (Bereich 1,5–19 Jahre) 1). Bei mehr als zwei Dritteln der berichteten Fälle wurde eine Bulbusperforation festgestellt 1). Bei mehr als der Hälfte der Fälle kommt es zu einem dauerhaften Sehverlust 1). Die blaue Sklera ist der häufigste Augenbefund und wurde bei 72 von 78 Fällen beobachtet 1).
System
Hauptbefunde
Häufigkeit
Auge
Hornhautverdünnung/-perforation, blaue Sklera
>90% 1)
Gelenke
Hypermobilität kleiner Gelenke
64/78 Fälle 1)
Hören
Schallempfindungs-, Schallleitungs- oder kombinierte Schwerhörigkeit
32/78 Fälle 1)
Gelenkhypermobilität ist der häufigste extraokuläre Befund 1). Angeborene Hüftdysplasie, Skoliose und Plattfüße wurden ebenfalls berichtet 1). Die Hörstörung betrifft tendenziell stärker die hohen Frequenzen 1). Eine Übercompliance des Trommelfells ist charakteristisch 1). Die Haut ist weich, überdehnbar und neigt zu Blutergüssen 1).
Berichte über Knochenbrüchigkeit haben in den letzten Jahren ebenfalls zugenommen. Zwei Brüder mit compound-heterozygoten ZNF469-Mutationen wiesen mehr als 10 Frakturen und Osteopenie auf 4). Es wird vermutet, dass etwa 16 % der BCS-Patienten eine Knochenbrüchigkeit aufweisen könnten 4). Die Knochenbiopsie zeigte eine Verdünnung der Kortikalis und eine deutliche Abnahme der kortikalen Porosität 4).
BCS ist eine autosomal-rezessive Erkrankung, die durch biallelische Mutationen in zwei Genen verursacht wird1).
ZNF469 ist an der normalen Entwicklung der Vorderkammer und der Hornhaut beteiligt1). Genomweite Assoziationsstudien haben einen Zusammenhang mit der zentralen Hornhautdicke gezeigt4). PRDM5 reguliert direkt die Transkription fibrillärer Kollagengene4). Funktionsverlustmutationen (Frame-Shift, Nonsense) sind in beiden Genen häufig1).
Heterozygote Mutationen in ZNF469 sind mit einer verringerten zentralen Hornhautdicke assoziiert2). Sie führen jedoch nicht zwangsläufig zu einer Hornhautverdünnung. Bei einem 64-jährigen chinesischen Mann mit heterozygoter ZNF469-Mutation war die Hornhautdicke normal (ca. 550 μm), es traten jedoch Hornhauttrübung und Epitheldefekt auf2).
Blutsverwandtschaft ist ein wichtiger Risikofaktor, der die Wahrscheinlichkeit homozygoter Mutationen erhöht1). In drei türkischen Fällen war die PRDM5-Mutation c.17T>G, p.(Val6Gly) gemeinsam, was auf einen möglichen Gründereffekt hindeutet1).
Die klinische Diagnose basiert auf einer globalen Hornhautverdünnung (zentrale Hornhautdicke <400 μm), blauen Skleren sowie systemischen Symptomen wie Gelenkhypermobilität und Hörverlust3). Hornhautpachymetrie und Hornhauttopographie mittels Pentacam sind unerlässlich3). Die OCT des vorderen Augenabschnitts ist für die detaillierte Beurteilung der Hornhautstruktur nützlich2)3).
Für die definitive Diagnose sind Gentests auf ZNF469 und PRDM5 erforderlich3). Die Exomsequenzierung (WES) ist effektiv zur Identifizierung pathogener Varianten3). Sie liefert auch nützliche Informationen für die genetische Beratung und Familienplanung3). Träger können Myopie oder eine leichte Hornhautverdünnung entwickeln2).
Bei BCS ist die Abgrenzung zum kyphoskoliotischen EDS (kEDS-PLOD1) besonders wichtig. Bei kEDS-PLOD1 reißt bei einer Augapfelruptur eher die Sklera als die Hornhaut1). Bei kEDS-PLOD1 sind Skoliose, Muskelhypotonie und vaskuläre Komplikationen ausgeprägter1).
QWarum ist die Frühdiagnose von BCS wichtig?
A
Das Durchschnittsalter für das Auftreten einer Hornhautperforation beträgt 4,3 Jahre, und die chirurgische Reparatur nach Perforation ist äußerst schwierig5). Bei mehr als der Hälfte der berichteten Fälle kommt es zu einem dauerhaften Sehverlust1). Eine Frühdiagnose ermöglicht das Tragen von Schutzbrillen und die Aufklärung zur Traumavermeidung1). Das Screening von Geschwistern ist ebenfalls wichtig für die Früherkennung5).
Der wichtigste Aspekt des BCS-Managements ist die Prävention einer Hornhautperforation1). Das ständige Tragen einer Schutzbrille aus Polycarbonat wird empfohlen5). Auch die prophylaktische Gabe von augendrucksenkenden Medikamenten wird in Betracht gezogen. Kontaktlinsen sind aufgrund der Hornhautverdünnung und des Traumarisikos eingeschränkt1).
Indikation: Durchgeführt bei schwerer Hornhautverdünnung oder zur Sehverbesserung nach Perforation3)
Operationsmethode: Verwendung eines 8,0-mm-Trephans, 16 unterbrochene 10-0-Nylonnähte3)
Ergebnisse: Bei albanischen Geschwistern verbesserte sich der bestkorrigierte Visus (BCVA) von 20/200 auf 20/30 und 20/253)
Verlauf: 7 Jahre ohne Komplikationen3)
Tiefe lamelläre Keratoplastik (DALK)
Vorteile: Erhalt des Endothels, dadurch geringeres Abstoßungsrisiko3)
Einschränkungen: Aufgrund der Fragilität des Stromas besteht ein intraoperatives Perforationsrisiko3)
Berichteter Fall: In einem Fall kam es intraoperativ zu einer zentralen Endothelperforation, die eine Konversion zur PKP erforderte3)
Auswahlkriterien: Ausreichende Stromadicke und Integrität der Descemet-Membran erforderlich3)
Bei der Reparatur von Hornhautperforationen stellt die extreme Gewebefragilität ein Problem dar. In einem Bericht über drei Brüder war die Verwendung langer Nahtbisse, Cyanacrylatkleber und eines Verbandskontaktlinsens wirksam5). Die sorgfältige Anlage eines korneoskleralen Tunnels kann Komplikationen reduzieren5). Bei einem 64-jährigen BCS-Patienten wurde eine PKP durchgeführt, der korrigierte Visus bei Entlassung betrug 0,22).
QIst das korneale Kollagen-Crosslinking wirksam?
A
Bei zwei pädiatrischen BCS-Patienten mit einer zentralen Hornhautdicke < 280 μm wurde ein transepitheliales CXL (mit Anpassung der UV-Dosis an die Hornhautdicke) durchgeführt3). Es wurde über eine Verbesserung des Visus und die Aufrechterhaltung der Endothelzelldichte berichtet3). Das Standard-Dresden-Protokoll erfordert jedoch eine zentrale Hornhautdicke ≥ 400 μm und ist bei extrem dünnen Hornhäuten < 200 μm kontraindiziert3). Protokollmodifikationen könnten den Anwendungsbereich erweitern, sind aber derzeit begrenzt.
ZNF469 und PRDM5 kodieren beide Transkriptionsfaktoren1). ZNF469 besitzt drei C2H2-Zinkfingerdomänen am C-Terminus1). Es reguliert die Expression extrazellulärer Matrixgene (CLU, GPC6, PCOLCE2, THBS1)1). PRDM5 besitzt eine PR-SET-Domäne und reguliert die Transkription von Kollagen-Genen direkt über die Bindung an RNA-Polymerase II4). Es ist auch an der Regulation des Wnt-Signalwegs beteiligt.
Diese Genmutationen stören die Kollagenablagerung und Faseranordnung im Hornhautstroma1). Die ZNF469-Mutation führt zu einer verminderten Expression von Kollagen Typ I (COL-I) und strukturellen Veränderungen 2). Die Immunfluoreszenzfärbung von Hornhautgewebe nach PKP zeigte eine Abnahme von COL-I und eine Zunahme von Kollagen Typ III 2). Auch die Masson-Färbung ergab eine deutliche Verringerung der Kollagenfasermenge 2).
Die konfokale Mikroskopie zeigt ein Netzwerk stark reflektierender linearer Strukturen im vorderen Hornhautstroma2). Es liegt keine Infiltration von Entzündungszellen vor 2). Dies deutet darauf hin, dass die Hornhautläsionen bei BCS auf eine primäre Anomalie der Kollagenstruktur zurückzuführen sind.
Auch im Knochengewebe bindet PRDM5 an die Exon-DNA des Kollagen-Typ-I-Gens 4). Die Knochenbiopsie zeigte eine Verdünnung der Kortikalis und eine deutliche Abnahme der kortikalen Porosität (1,3 %; Normalwert 6,3 ± 0,6 %) 4). Ein Panel von 28 Genen für Knochenbrüchigkeit ergab keine weiteren pathogenen Mutationen, was darauf hindeutet, dass die ZNF469-Mutation selbst die Ursache für die Knochenbrüchigkeit sein könnte 4).
QWarum sind bei BCS auch andere Gewebe als die Hornhaut betroffen?
A
ZNF469 und PRDM5 sind an der transkriptionellen Regulation der extrazellulären Matrix im gesamten Bindegewebe beteiligt 1). Die strukturellen Anomalien der Kollagenfasern beschränken sich nicht auf die Hornhaut; sie betreffen das Bindegewebe im ganzen Körper, einschließlich der Sklera (blaue Verfärbung), Gelenke (Hypermobilität), Haut (Überdehnbarkeit), Knochen (Osteopenie) und Trommelfell (Übercompliance) 4). Berichte über Osteopenie und Frakturen deuten darauf hin, dass BCS einen Phänotyp der Knochenbrüchigkeit aufweisen könnte 4).
Fortschritte in der Gentherapie eröffnen neue Möglichkeiten für BCS. CRISPR-Genom-Editierung und RNA-Interferenz (RNAi) werden als vielversprechende Behandlungen zur Stabilisierung oder Verbesserung der Hornhautverdünnung angesehen 3).
Biotechnologisch hergestellte Hornhautimplantate (BPCDX) sind zellfreie transparente Hydrogele aus medizinischem Schweinekollagen 3). Es handelt sich um ein minimalinvasives Verfahren, bei dem das Implantat in eine 2–3 mm große intrastromale Tasche eingesetzt wird, die mit einem Femtosekundenlaser erzeugt wurde 3). Eine 24-monatige Beobachtung ergab eine Zunahme der Hornhautdicke, eine Abflachung der maximalen Keratometrie um 18 D und eine Verbesserung der korrigierten Sehschärfe3). Stabile Ergebnisse wurden ohne Abstoßung erzielt 3).
Auch die intrastromale Injektion mesenchymaler Stammzellen wird untersucht 3). Stammzellen aus Fettgewebe und Knochenmark werden in einen lamellären Schnitt injiziert, der mit einem Femtosekundenlaser erzeugt wurde, was eine Verzögerung der PKP ermöglichen könnte 3). Derzeit wird diese Methode nur bei Patienten mit Keratokonus angewendet, aber eine Anwendung bei BCS-Patienten wird erwartet 3).
Sanri A, Demir S, Gürkan H. Homozygous Val6Gly Variation in PRDM5 Gene Causing Brittle Cornea Syndrome: A New Turkish Case. Mol Syndromol. 2023;14(2):129-135. doi:10.1159/000524832. PMID:37064337; PMCID:PMC10091010.
Geng X, Zhu L, Li J, Li Z. Brittle cornea syndrome: A novel mutation. Heliyon. 2024;10:e32506. doi:10.1016/j.heliyon.2024.e32506.
Zeppieri M, Gentile M, Acquaviva A, Scollo D, D’Esposito F, Gagliano G, et al. Brittle Cornea Syndrome: Molecular Diagnosis and Management. Diagnostics (Basel, Switzerland). 2025;15(13). doi:10.3390/diagnostics15131596. PMID:40647596; PMCID:PMC12249002.
Cundy T, Vincent A, Robertson S. Does brittle cornea syndrome have a bone fragility phenotype? Bone Rep. 2021;15:101124. doi:10.1016/j.bonr.2021.101124. PMID:34522702; PMCID:PMC8426531.
Mandlik K, Betdur RA, Rashmita R, Narayana S. Brittle cornea syndrome: A tale of three brothers. Indian journal of ophthalmology. 2022;70(7):2594-2597. doi:10.4103/ijo.IJO_2894_21. PMID:35791165; PMCID:PMC9426125.
Kopieren Sie den Artikeltext und fügen Sie ihn in den KI-Assistenten Ihrer Wahl ein.
Artikel in die Zwischenablage kopiert
Öffnen Sie unten einen KI-Assistenten und fügen Sie den kopierten Text in den Chat ein.