Le neuroblastome (NB) est une tumeur maligne qui se développe à partir des cellules progénitrices du système nerveux sympathique dérivées de la crête neurale. Il représente environ 10 % des tumeurs malignes de l’enfant et est la tumeur solide extracrânienne la plus fréquente chez l’enfant. C’est également la deuxième tumeur maligne la plus fréquente après la leucémie.
Le site primitif est la glande surrénale dans environ 46 % des cas, l’abdomen (extra-surrénalien) dans environ 18 %, le médiastin postérieur dans environ 14 %, et le bassin/autres dans environ 22 % des cas4). Plus de 60 % des cas surviennent dans l’abdomen, au niveau des glandes surrénales ou des ganglions sympathiques.
Caractéristiques épidémiologiques :
Environ 700 cas sont diagnostiqués chaque année aux États-Unis
L’âge médian au diagnostic est de 19 mois. 90 % des cas sont diagnostiqués avant l’âge de 5 ans5)
L’incidence chez l’adulte est extrêmement rare, avec 1 cas par an pour 10 millions de personnes5)
Sur le plan ophtalmologique, 11 à 56 % des neuroblastomes métastasent à l’orbite. Le tableau clinique est varié, allant de la régression spontanée aux métastases étendues.
Contexte historique :
1864 : Virchow rapporte pour la première fois une tumeur abdominale comme un « gliome »
1891 : Marchand décrit les caractéristiques dérivées de la médullosurrénale et du système nerveux sympathique
1901 : Pepper rapporte un cas de nourrisson avec métastases hépatiques (correspondant au stade MS actuel)
1910 : Homer Wright décrit les pseudorosettes de Homer-Wright dans la moelle osseuse
QOù le neuroblastome métastase-t-il fréquemment ?
A
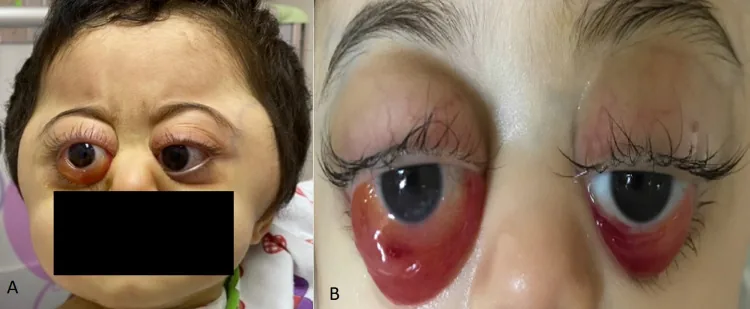
Les métastases sont fréquentes dans les os, la moelle osseuse, les ganglions lymphatiques, le foie et la peau. La fréquence des métastases orbitaires est de 11 à 56 %, et une exophtalmie ou des ecchymoses périorbitaires (yeux de raton laveur) peuvent être les premiers symptômes.
Rahaf A Mandura. Rapidly Progressive Ocular Proptosis as the First Sign of Neuroblastoma in a 16-Month-Old Child: Case Report and Review of Literature. Cureus. 2022 Jan 6; 14(1):e20982. Figure 1. PMCID: PMC8817620. License: CC BY.
Les signes cliniques se divisent en signes ophtalmologiques liés à une métastase orbitaire et en signes neurologiques dus à la tumeur.
Signes orbitaires et périorbitaires
Hématome périorbitaire (yeux de raton laveur) : signe le plus fréquent des métastases orbitaires chez les enfants de moins de 2 ans. Il est dû à une nécrose hémorragique de la tumeur. Il est souvent confondu avec des sévices.
Exophtalmie (proptosis) : déplacement antérieur du globe oculaire dû à la masse tumorale intraorbitaire.
Œdème palpébral et conjonctival : l’œdème peut devenir marqué.
Ecchymose palpébrale : signe caractéristique du NB et de la leucémie.
Destruction osseuse orbitaire : modifications ostéolytiques confirmées par imagerie.
Signes neurologiques et du fond d'œil
Syndrome de Horner : triade myosis, ptosis, anhidrose. Survient lorsque la tumeur thoracique primitive implique la chaîne sympathique.
Hétérochromie irienne : observée dans le syndrome de Horner dû à un NB ganglionnaire cervical congénital.
Opsoclonus : mouvements oculaires saccadés de haute fréquence (10-15 Hz) et multidirectionnels. Associé à des myoclonies.
Paralysie du droit externe, strabisme, ésotropie : dus à un trouble de la motilité oculaire.
Œdème papillaire, hémorragie rétinienne, atrophie optique : dus à une hypertension intracrânienne ou à une infiltration directe.
QSi un enfant présente des ecchymoses autour des yeux, est-ce que cela pourrait être un neuroblastome ?
A
L’hémorragie sous-cutanée périorbitaire (yeux de raton laveur) est le signe le plus fréquent des métastases orbitaires chez les enfants de moins de 2 ans. Une hémorragie périorbitaire bilatérale sans antécédent de traumatisme doit faire suspecter activement un neuroblastome. Comme elle peut être confondue avec des sévices, un bilan complet incluant une échographie abdominale et un dosage des catécholamines urinaires est nécessaire pour le diagnostic différentiel.
QQu'est-ce que le syndrome opsoclonus-myoclonus ?
A
Il s’agit d’un syndrome associant des mouvements oculaires saccadiques multidirectionnels à haute fréquence (10-15 Hz) et un myoclonus. Il est considéré comme un syndrome paranéoplasique dû à des anticorps anormaux dirigés contre l’ARN neuronal. Un neuroblastome occulte est présent chez 48 % des patients atteints d’OMS, ce qui rend un bilan complet obligatoire chez l’enfant.
La majorité des cas sont sporadiques, sans facteur de risque clairement identifié.
Facteurs génétiques (environ 1 à 2 % des cas) :
Mutation germinale d’ALK : cause la plus fréquente du NB familial
Anomalie de PHOX2B : associée à un trouble de la différenciation en neurones matures
Anomalie de KIF1B / amplification de MYCN : une partie du NB familial
Polymorphisme du nombre de copies de NBPF10 (délétions 1p/11q) : associé au NB à haut risque
Duplication du gène LMO1 : facteur de risque de NB agressif
Association avec NF1 et syndrome de Beckwith-Wiedemann : rarement associés
Mutations génétiques associées au NB sporadique :
Variation génétique en 6p22 : associée au NB sporadique
L’amplification de MYCN est observée dans 20 à 25 % des NB de l’enfant et présente une distribution bimodale (3 à 10 fois ou 100 à 300 fois). C’est un marqueur moléculaire fortement lié aux NB agressifs5).
Caractéristiques moléculaires du NB de l’adulte : différentes de celles de l’enfant, l’amplification de MYCN est rare. Les mutations d’ATRX (11 %), d’ALK (jusqu’à 14 %) et la réorganisation de TERT (23 %) sont caractéristiques5).
Facteurs de risque périnatals (d’après des études de cohorte rétrospectives) :
Nourrissons de faible âge : poids de naissance élevé, prise de poids excessive de la mère, hypertension gravidique, âge maternel avancé.
Nourrissons âgés : faible poids de naissance augmente le risque
Tranche d’âge élevée : premier enfant, première césarienne, travail prolongé, rupture prématurée des membranes
Utilisation de médicaments par la mère (diurétiques, antihypertenseurs), anémie pendant la grossesse, utilisation temporaire de teinture capillaire
Aucun facteur de risque spécifique aux métastases orbitaires n’est connu.
Catécholamines urinaires (HVA·VMA) : auparavant rapportées comme élevées dans 90 à 95 % des cas, mais des études récentes montrent une sensibilité plus faible. Chez l’adulte, elle n’est que de 40 à 57 % (contre 95 % chez l’enfant)
Analyses sanguines : pancytopénie, fonctions hépatique et rénale, électrolytes, ferritine, LDH, fonction thyroïdienne
Biopsie médullaire : évaluation de l’infiltration médullaire par aspiration et biopsie osseuse des crêtes iliaques postérieures bilatérales
Tests d’anticorps : l’anticorps anti-Hu (ANNA-1) est utile pour le diagnostic du neuroblastome chez l’enfant
Ponction lombaire : à éviter sauf si une métastase du SNC est déjà connue (des cas d’augmentation des métastases du SNC après PL ont été rapportés)
Hypersignal T1, hétérogène T2, évaluation de l’extension intracrânienne
Examen détaillé intracrânien et orbitaire
MIBG (123I)
Idéal pour identifier les métastases des tissus mous et osseuses
Évaluation systémique des métastases
TEP
Haute sensibilité et spécificité
Diagnostic et suivi thérapeutique
La scintigraphie MIBG (123I-MIBG) est supérieure à la TEP/TDM pour l’identification des métastases des tissus mous et osseuses. Dans les métastases orbitaires, la TDM montre des changements ostéolytiques, et l’IRM montre un signal isointense par rapport aux muscles extra-oculaires en T1, un signal plus faible que la graisse, et un signal plus élevé que les muscles extra-oculaires et la graisse en T2. Les métastases sont fréquentes dans la paroi postéro-latérale de l’orbite.
QQuel est l'examen d'imagerie le plus utile pour le neuroblastome ?
A
La scintigraphie MIBG (123I-MIBG) est la plus utile pour l’identification des métastases des tissus mous et osseuses, et est considérée comme supérieure à la TEP/TDM. Comme elle permet d’évaluer l’ensemble des métastases en une seule fois, elle est utilisée à la fois pour la stadification initiale et pour l’évaluation de la réponse au traitement.
L’exérèse complète est rarement indiquée pour les tumeurs orbitaires métastatiques. On administre un traitement efficace contre le cancer primitif, comme la chimiothérapie ou la radiothérapie. Chez les enfants atteints de neuroblastome (NB) qui répondent à la chimiothérapie, le pronostic vital est relativement bon. En présence de métastases orbitaires, le taux de survie à 5 ans est très faible, à 7,6 %.
Il n’existe pas de protocole standard établi ; on adapte souvent le protocole pédiatrique5). Une chirurgie (y compris laparoscopique) associée à une chimiothérapie (carboplatine + étoposide, etc.) est réalisée. Le taux de survie globale à 5 ans chez les adultes de 20 ans et plus est de 36,3 %, ce qui est de mauvais pronostic5).
QQuel est le pronostic lorsque le neuroblastome métastase à l'orbite ?
A
Le taux de survie à 5 ans du neuroblastome avec métastases orbitaires est très faible, à 7,6 %. Les métastases orbitaires indiquent souvent une maladie métastatique à distance (équivalent au stade M), et un traitement multimodal est administré en tant que groupe à haut risque, mais l’amélioration du pronostic est limitée.
Le NB provient des précurseurs du système nerveux sympathique (sympathoblastes). La mutation PHOX2B entraîne une altération de la différenciation en neurones matures, conduisant à la tumorigenèse. Les mutations ALK sont associées à une diminution de la prolifération et à une augmentation des neurones sympathiques immatures.
Anomalie du métabolisme des catécholamines : En raison d’un défaut de synthèse des catécholamines dans les cellules tumorales, les métabolites intermédiaires HVA et VMA s’accumulent et sont excrétés dans l’urine. C’est la base du test urinaire des catécholamines.
L’activation de la voie des polyamines par l’amplification de MYCN favorise la croissance tumorale3).
MYCN (facteur de transcription de la famille MYC) → Augmentation de la transcription d’ODC1 → Promotion de la synthèse des polyamines (ornithine → putrescine → spermidine → spermine) → Activation d’eIF5A → Activation de LIN28 → Suppression du miRNA Let-7 → Promotion de la croissance tumorale
Syndrome opsoclonus-myoclonus (OMS) : causé par des anticorps anormaux dirigés contre l’ARN des neurones. Considéré comme un symptôme paranéoplasique dû à une réaction croisée avec les tissus.
Mécanisme des troubles visuels (sans compression directe) : la réaction immunitaire contre le NB réagit de manière croisée avec les tissus. Les métabolites toxiques dérivés du cancer et l’arrêt du transport axonal dû à la chimiothérapie peuvent également être impliqués.
Certains NB (en particulier le stade MS) présentent une régression spontanée. Les mécanismes impliqueraient une hyperméthylation de l’ADN sous-télomérique, l’apoptose, une carence en facteur de croissance nerveuse (NGF) et une réaction immunitaire.
Caractéristiques moléculaires et biologiques du NB chez l’enfant vs l’adulte
Les caractéristiques génétiques diffèrent considérablement entre les enfants et les adultes1)5).
Caractéristique
NB pédiatrique
NB adulte
Amplification de MYCN
20 à 25 %
Rare
Expression de PHOX2B
Élevée (haute sensibilité et spécificité)
50% négatifs
Mutation ATRX
Rare
11%
Mutation ALK
Rare
Jusqu’à 14 %
Réarrangement TERT
Rare
23 %
Le fait que 50% des NB adultes soient PHOX2B négatifs suggère une lignée cellulaire différente (par exemple, d’origine thymique) par rapport au type pédiatrique 1).
7. Recherches récentes et perspectives futures (rapports en phase de recherche)
Le 13 décembre 2023, la FDA américaine a approuvé un nouveau médicament pour le traitement d’entretien du NB à haut risque 3).
Jiang & Yu et al. (2024) ont détaillé le profil pharmacologique de l’éflornithine, un inhibiteur irréversible (suicide inhibitor) de l’ODC (ornithine décarboxylase), ciblant la voie des polyamines dans le NB avec amplification de MYCN 3). L’essai de phase 2 (NCT02395666) en tant que traitement d’entretien après immunothérapie (dinutuximab) est terminé, conduisant à l’approbation de la FDA.
Propriétés du médicament :
Masse moléculaire : 182,2 g/mol
Voie d’administration : orale
Demi-vie : environ 3,5 heures (excrétion rénale, pratiquement pas métabolisé)
Indication : traitement d’entretien après immunothérapie pour le neuroblastome à haut risque
Des stratégies de traitement personnalisées utilisant la planification thérapeutique numérique (Oncompass™, etc.) sont à l’étude5).
QQu'est-ce que l'éflornithine (IWILFIN) ?
A
C’est un médicament oral approuvé par la FDA en décembre 2023 pour le traitement d’entretien après immunothérapie du neuroblastome à haut risque. Il inhibe de manière irréversible l’ODC (ornithine décarboxylase), supprimant ainsi la synthèse des polyamines et bloquant les signaux de croissance tumorale induits par l’amplification de MYCN3). Il est nécessaire de vérifier auprès du médecin traitant le statut d’approbation au Japon.
Collins K, Ulbright TM, Davis JL. Anterior mediastinal neuroblastoma in an adult: an additional case of a rare tumor in an unusual location with review of the literature. Diagn Pathol. 2023;18:127.
Hu J, Xia B, Yuan X, et al. Neuroblastoma with superficial soft tissue mass as the first symptom: case reports with atypical ultrasonic image and literature review. Braz J Med Biol Res. 2023;56:e12975.
Jiang J, Yu Y. Eflornithine for treatment of high-risk neuroblastoma. Trends Pharmacol Sci. 2024;45(6):577-578.
do Amaral-Silva GK, Leite AA, Mariz BALA, et al. Metastatic neuroblastoma to the mandible of children: report of two cases and critical review of the literature. Head Neck Pathol. 2021;15:757-768.
Telecan T, Andras I, Bungardean MR, et al. Adrenal gland primary neuroblastoma in an adult patient: a case report and literature review. Medicina. 2023;59:33.
Copiez le texte de l'article et collez-le dans l'assistant IA de votre choix.
Article copié dans le presse-papiers
Ouvrez un assistant IA ci-dessous et collez le texte copié dans la conversation.