Das Neuroblastom (NB) ist ein bösartiger Tumor, der aus Vorläuferzellen des sympathischen Nervensystems entsteht, die von der Neuralleiste abstammen. Es macht etwa 10 % der bösartigen Tumoren im Kindesalter aus und ist der häufigste extrakranielle solide Tumor bei Kindern. Es ist auch der zweithäufigste bösartige Tumor nach Leukämie.
Der primäre Ursprungsort ist in etwa 46 % der Fälle die Nebenniere, in etwa 18 % das Abdomen (extradrenal), in etwa 14 % das hintere Mediastinum und in etwa 22 % das Becken/Sonstiges4). Mehr als 60 % treten im Abdomen auf, entweder in den Nebennieren oder in den sympathischen Ganglien.
Epidemiologische Merkmale sind wie folgt:
In den USA werden jährlich etwa 700 Fälle diagnostiziert
Das mediane Alter bei Diagnose beträgt 19 Monate. 90 % werden vor dem 5. Lebensjahr diagnostiziert5)
Das Auftreten bei Erwachsenen ist extrem selten, mit 1 Fall pro 10 Millionen Menschen pro Jahr5)
Augenärztlich gesehen metastasieren 11–56 % der Neuroblastome in die Orbita. Das klinische Bild reicht von spontaner Regression bis hin zu ausgedehnten Metastasen.
Historischer Hintergrund:
1864: Virchow berichtet erstmals über einen abdominalen Tumor als „Gliom“
1891: Marchand beschreibt die Merkmale des Ursprungs aus dem Nebennierenmark und dem sympathischen Nervensystem
1901: Pepper berichtet über einen Säuglingsfall mit Lebermetastasen (entspricht dem heutigen Stadium MS)
1910: Homer Wright beschreibt die Homer-Wright-Pseudorosetten im Knochenmark
QWohin metastasiert das Neuroblastom häufig?
A
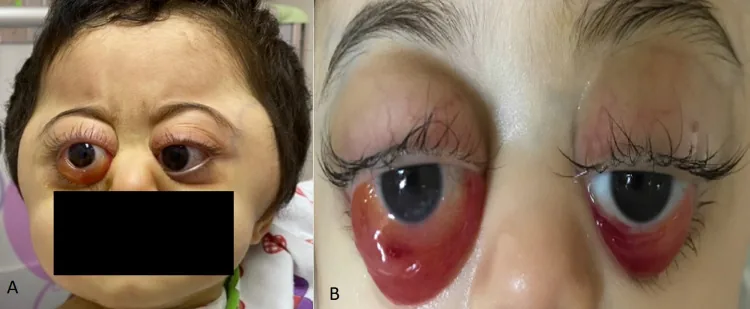
Metastasen treten häufig in Knochen, Knochenmark, Lymphknoten, Leber und Haut auf. Die Häufigkeit von Orbita-Metastasen beträgt 11–56 %, und Exophthalmus oder periorbitale Blutergüsse (Waschbäraugen) können die ersten Symptome sein.
Rahaf A Mandura. Rapidly Progressive Ocular Proptosis as the First Sign of Neuroblastoma in a 16-Month-Old Child: Case Report and Review of Literature. Cureus. 2022 Jan 6; 14(1):e20982. Figure 1. PMCID: PMC8817620. License: CC BY.
Die klinischen Befunde werden unterteilt in ophthalmologische Befunde im Zusammenhang mit einer orbitalen Metastase und neurologische Befunde durch den Tumor.
Orbitale und periorbitale Befunde
Periorbitales Hämatom (Waschbärenaugen): Häufigster Befund bei orbitalen Metastasen unter 2 Jahren. Beruht auf hämorrhagischer Nekrose des Tumors. Wird oft mit Misshandlung verwechselt.
Exophthalmus (Proptosis): Vorwärtsverlagerung des Augapfels durch die intraorbitale Tumormasse.
Lid- und Bindehautödem: Das Ödem kann ausgeprägt sein.
Lidekchymose: Charakteristischer Befund bei NB und Leukämie.
Orbitale Knochenzerstörung: Osteolytische Veränderungen in der Bildgebung.
Neurologische und Fundusbefunde
Horner-Syndrom: Trias aus Miosis, Ptosis, Anhidrose. Tritt auf, wenn der thorakale Primärtumor die sympathische Kette einbezieht.
Iris-Heterochromie: Bei Horner-Syndrom durch kongenitales zervikales Ganglioneuroblastom.
Lateralis-Parese, Strabismus, Esotropie : aufgrund einer Augenbewegungsstörung.
Stauungspapille, Netzhautblutung, Optikusatrophie : durch erhöhten intrakraniellen Druck oder direkte Infiltration.
QKann ein Bluterguss um die Augen eines Kindes auf ein Neuroblastom hinweisen?
A
Periorbitale subkutane Blutungen (Raccoon Eyes) sind der häufigste Befund bei orbitalen Metastasen bei Kindern unter 2 Jahren. Bei beidseitigen periorbitalen Blutungen ohne Trauma-Anamnese sollte aktiv an ein Neuroblastom gedacht werden. Da sie leicht mit Misshandlung verwechselt werden können, ist zur Differenzialdiagnose eine umfassende Untersuchung einschließlich abdominalem Ultraschall und Urin-Katecholamin-Test erforderlich.
QWas ist das Opsoklonus-Myoklonus-Syndrom?
A
Es handelt sich um ein Syndrom mit hochfrequenten (10–15 Hz), multidirektionalen sakkadischen Augenbewegungen, begleitet von Myoklonus. Es wird als paraneoplastisches Syndrom aufgrund abnormaler Antikörper gegen neuronale RNA angesehen. Bei 48 % der OMS-Patienten liegt ein okkultes Neuroblastom vor, sodass bei Kindern mit OMS eine vollständige Untersuchung obligat ist.
Die meisten Fälle sind sporadisch, ohne dass ein eindeutiger Risikofaktor identifiziert wurde.
Genetische Faktoren (ca. 1–2 % aller Fälle):
ALK-Keimbahnmutation : häufigste Ursache des familiären NB
PHOX2B-Anomalie : assoziiert mit gestörter Differenzierung zu reifen Neuronen
KIF1B-Anomalie / MYCN-Amplifikation : Teil des familiären NB
NBPF10-Kopienzahlpolymorphismus (1p/11q-Deletionen) : assoziiert mit Hochrisiko-NB
LMO1-Genduplikation : Risikofaktor für aggressives NB
Assoziation mit NF1 und Beckwith-Wiedemann-Syndrom : selten gemeinsam auftretend
Genmutationen im Zusammenhang mit sporadischem NB:
Genetische Variante auf 6p22: mit sporadischem NB assoziiert
MYCN-Amplifikation tritt bei 20–25 % der pädiatrischen NB auf und zeigt eine bimodale Verteilung (3- bis 10-fach oder 100- bis 300-fach). Sie ist ein starker molekularer Marker für aggressives NB5).
Molekularbiologische Merkmale des adulten NB unterscheiden sich von denen des pädiatrischen NB; MYCN-Amplifikation ist selten. Charakteristisch sind ATRX-Mutationen (11 %), ALK-Mutationen (bis zu 14 %) und TERT-Rearrangements (23 %)5).
Urin-Katecholamine (HVA·VMA) : Früher wurde bei 90–95 % eine Erhöhung berichtet, aber neuere Studien zeigen eine geringere Sensitivität. Bei Erwachsenen liegt sie nur bei 40–57 % (im Gegensatz zu 95 % bei Kindern)
Blutuntersuchungen : Panzytopenie, Leber- und Nierenfunktion, Elektrolyte, Ferritin, LDH, Schilddrüsenfunktion
Knochenmarkbiopsie : Beurteilung der Knochenmarkinfiltration durch Aspiration und Stanzbiopsie von beiden hinteren Beckenkämmen
Antikörpertests : Anti-Hu-Antikörper (ANNA-1) sind für die Diagnose des kindlichen Neuroblastoms nützlich
Lumbalpunktion: Vermeidung außer bei bekannter ZNS-Metastasierung (Berichte über erhöhte ZNS-Metastasierung nach LP)
T1-hypointens, T2-inhomogen, Beurteilung der intrakraniellen Ausdehnung
Intrakranielle und orbitale Detailuntersuchung
MIBG (123I)
Optimal zur Identifizierung von Weichteil- und Knochenmetastasen
Ganzkörperbeurteilung von Metastasen
PET
Hohe Sensitivität und Spezifität
Diagnose und Therapieüberwachung
Die MIBG-Szintigraphie (123I-MIBG) ist der PET/CT bei der Identifizierung von Weichteil- und Knochenmetastasen überlegen. Bei orbitalen Metastasen zeigt die CT osteolytische Veränderungen, und die MRT zeigt in T1 ein isointenses Signal zu den äußeren Augenmuskeln und ein niedrigeres Signal als Fett, in T2 ein höheres Signal als die äußeren Augenmuskeln und Fett. Metastasen treten häufig an der posterolateralen Wand der Orbita auf.
QWelche bildgebende Untersuchung ist beim Neuroblastom am nützlichsten?
A
Die MIBG-Szintigraphie (123I-MIBG) ist die nützlichste Methode zur Identifizierung von Weichteil- und Knochenmetastasen und gilt als der PET/CT überlegen. Da sie alle Metastasen auf einmal beurteilen kann, wird sie sowohl für das initiale Staging als auch für die Beurteilung des Therapieansprechens eingesetzt.
Eine vollständige Entfernung ist bei metastasierten Orbitatumoren selten indiziert. Es wird eine wirksame Behandlung des Primärtumors wie Chemotherapie oder Strahlentherapie durchgeführt. Bei Kindern mit Neuroblastom (NB), die auf eine Chemotherapie ansprechen, ist die Lebensprognose relativ gut. Bei orbitalen Metastasen beträgt die 5-Jahres-Überlebensrate 7,6 %, was sehr schlecht ist.
Es gibt kein etabliertes Standardprotokoll; häufig wird das pädiatrische Protokoll angepasst5). Eine Operation (einschließlich laparoskopisch) + Chemotherapie (Carboplatin + Etoposid usw.) wird durchgeführt. Die 5-Jahres-Gesamtüberlebensrate bei Erwachsenen ab 20 Jahren beträgt 36,3 %, was eine schlechte Prognose darstellt5).
QWie ist die Prognose, wenn ein Neuroblastom in die Augenhöhle metastasiert?
A
Die 5-Jahres-Überlebensrate bei Neuroblastom mit orbitalen Metastasen beträgt 7,6 % und ist damit sehr schlecht. Orbitale Metastasen deuten oft auf eine Fernmetastasierung (entspricht Stadium M) hin, und es wird eine multimodale Therapie als Hochrisikogruppe durchgeführt, aber die Prognoseverbesserung ist begrenzt.
6. Pathophysiologie und detaillierter Pathomechanismus
NB entsteht aus Vorläuferzellen des sympathischen Nervensystems (Sympathoblasten). Die PHOX2B-Mutation beeinträchtigt die Differenzierung zu reifen Neuronen, was zur Tumorentstehung führt. ALK-Mutationen sind mit verminderter Proliferation und einer Zunahme unreifer sympathischer Neuronen verbunden.
Störung des Katecholaminstoffwechsels: Aufgrund eines Defekts der Katecholaminsynthese in Tumorzellen akkumulieren die Zwischenmetaboliten HVA und VMA und werden im Urin ausgeschieden. Dies ist die Grundlage des Urin-Katecholamin-Tests.
Opsoklonus-Myoklonus-Syndrom (OMS) : Verursacht durch abnorme Antikörper gegen neuronale RNA. Es wird als paraneoplastisches Symptom aufgrund von Kreuzreaktionen mit Gewebe angesehen.
Mechanismus der Sehstörung (ohne direkte Kompression) : Die Immunreaktion gegen NB kreuzreagiert mit Gewebe. Auch krebsbedingte toxische Metaboliten und durch Chemotherapie verursachter Stillstand des axonalen Transports können beteiligt sein.
Einige NB (insbesondere Stadium MS) zeigen eine spontane Regression. Als Mechanismen werden Hypermethylierung subtelomerer DNA, Apoptose, Mangel an Nervenwachstumsfaktor (NGF) und Immunreaktionen diskutiert.
Molekularbiologische Merkmale von NB bei Kindern vs. Erwachsenen
Am 13. Dezember 2023 hat die US-amerikanische FDA ein neues Medikament als Erhaltungstherapie für Hochrisiko-NB zugelassen 3).
Jiang & Yu et al. (2024) beschrieben das pharmakologische Profil von Eflornithin, einem irreversiblen Inhibitor (Suizid-Inhibitor) der ODC (Ornithindecarboxylase), der den Polyaminweg in MYCN-amplifiziertem NB angreift 3). Die Phase-2-Studie (NCT02395666) als Erhaltungstherapie nach Immuntherapie (Dinutuximab) wurde abgeschlossen, was zur FDA-Zulassung führte.
Wirkstoffeigenschaften:
Molekulargewicht: 182,2 g/mol
Verabreichungsweg: oral
Halbwertszeit: ca. 3,5 Stunden (renale Ausscheidung, nahezu kein Metabolismus)
Indikation: Erhaltungstherapie nach Immuntherapie bei Hochrisiko-Neuroblastom
Personalisierte Behandlungsstrategien unter Verwendung digitaler Therapieplanung (Oncompass™ usw.) werden untersucht5).
QWas ist Eflornithin (IWILFIN) für ein Medikament?
A
Es ist ein orales Medikament, das im Dezember 2023 von der FDA zur Erhaltungstherapie nach Immuntherapie des Hochrisiko-Neuroblastoms zugelassen wurde. Es hemmt irreversibel die ODC (Ornithindecarboxylase), unterdrückt die Polyaminsynthese und blockiert die durch MYCN-Amplifikation verursachten Tumorwachstumssignale3). Der Zulassungsstatus in Japan muss mit dem behandelnden Arzt geklärt werden.
Collins K, Ulbright TM, Davis JL. Anterior mediastinal neuroblastoma in an adult: an additional case of a rare tumor in an unusual location with review of the literature. Diagn Pathol. 2023;18:127.
Hu J, Xia B, Yuan X, et al. Neuroblastoma with superficial soft tissue mass as the first symptom: case reports with atypical ultrasonic image and literature review. Braz J Med Biol Res. 2023;56:e12975.
Jiang J, Yu Y. Eflornithine for treatment of high-risk neuroblastoma. Trends Pharmacol Sci. 2024;45(6):577-578.
do Amaral-Silva GK, Leite AA, Mariz BALA, et al. Metastatic neuroblastoma to the mandible of children: report of two cases and critical review of the literature. Head Neck Pathol. 2021;15:757-768.
Telecan T, Andras I, Bungardean MR, et al. Adrenal gland primary neuroblastoma in an adult patient: a case report and literature review. Medicina. 2023;59:33.
Kopieren Sie den Artikeltext und fügen Sie ihn in den KI-Assistenten Ihrer Wahl ein.
Artikel in die Zwischenablage kopiert
Öffnen Sie unten einen KI-Assistenten und fügen Sie den kopierten Text in den Chat ein.