El neuroblastoma (NB) es un tumor maligno que se origina a partir de células precursoras del sistema nervioso simpático derivadas de la cresta neural. Representa aproximadamente el 10% de los tumores malignos infantiles y es el tumor sólido extracraneal más común en niños. También es el segundo tumor maligno más frecuente después de la leucemia.
Los sitios primarios incluyen la glándula suprarrenal en aproximadamente el 46% de los casos, el abdomen (extra-suprarrenal) en aproximadamente el 18%, el mediastino posterior en aproximadamente el 14%, y la pelvis/otros en aproximadamente el 22% 4). Más del 60% ocurren en el abdomen, como la glándula suprarrenal o los ganglios simpáticos.
Las características epidemiológicas son las siguientes:
Aproximadamente 700 casos se diagnostican anualmente en los Estados Unidos
La mediana de edad al diagnóstico es de 19 meses. El 90% se diagnostica antes de los 5 años 5)
La aparición en adultos es extremadamente rara, con 1 caso por cada 10 millones de personas al año 5)
Desde el punto de vista oftalmológico, entre el 11% y el 56% de los neuroblastomas metastatizan en la órbita. El cuadro clínico es variado, desde regresión espontánea hasta metástasis extensa.
Antecedentes históricos son los siguientes:
1864: Virchow reportó por primera vez un tumor abdominal como “glioma”
1891: Marchand describió características originadas en la médula suprarrenal y el sistema nervioso simpático
1901: Pepper reportó casos infantiles con metástasis hepática (equivalente al estadio MS actual)
1910: Homer Wright describió pseudorrosetas de Homer-Wright en la médula ósea
Q¿Dónde suele metastatizar el neuroblastoma?
A
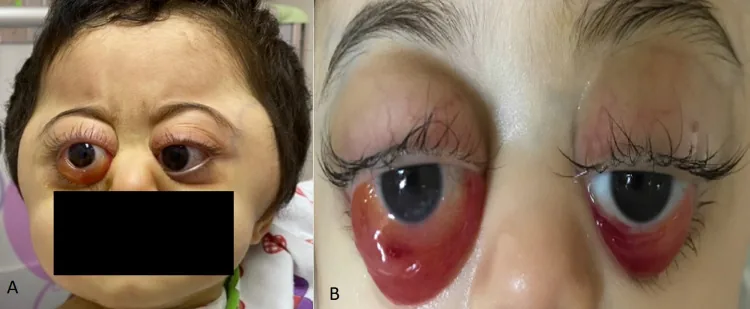
Las metástasis son frecuentes en huesos, médula ósea, ganglios linfáticos, hígado y piel. La metástasis orbitaria ocurre en un 11–56% de los casos, y la proptosis o equimosis periorbitaria (ojos de mapache) pueden ser el síntoma inicial.
Rahaf A Mandura. Rapidly Progressive Ocular Proptosis as the First Sign of Neuroblastoma in a 16-Month-Old Child: Case Report and Review of Literature. Cureus. 2022 Jan 6; 14(1):e20982. Figure 1. PMCID: PMC8817620. License: CC BY.
Se dividen en hallazgos oftalmológicos asociados con metástasis orbitaria y hallazgos neurológicos debidos al tumor.
Hallazgos orbitarios y periorbitarios
Equimosis periorbitaria (ojos de mapache): El hallazgo más común en metástasis orbitaria en niños menores de 2 años. Se debe a necrosis hemorrágica del tumor. Se confunde fácilmente con maltrato infantil.
Proptosis: Desplazamiento anterior del ojo debido a una masa tumoral intraorbitaria.
Edema palpebral y conjuntival: El edema puede volverse prominente.
Equimosis palpebral: Un hallazgo característico en NB y leucemia.
Destrucción ósea orbitaria: Cambios osteolíticos confirmados por imágenes.
Hallazgos neurológicos y de fondo de ojo
Síndrome de Horner: Tríada de miosis, ptosis y anhidrosis. Ocurre cuando un tumor torácico primario afecta la cadena simpática.
Heterocromía del iris: Se observa en el síndrome de Horner debido a NB del ganglio cervical congénito.
Opsoclonus: Movimientos oculares sacádicos de alta frecuencia (10-15 Hz) y multidireccionales. Acompañado de mioclonías.
Parálisis del recto lateral, estrabismo, esotropía: Debido a la alteración del movimiento ocular.
Edema de papila, hemorragia retiniana, atrofia óptica: Debido al aumento de la presión intracraneal o infiltración directa.
QSi un niño tiene moretones alrededor de los ojos, ¿podría ser neuroblastoma?
A
La hemorragia subcutánea periorbitaria (ojos de mapache) es el hallazgo más común en la metástasis orbitaria en niños menores de 2 años. La hemorragia periorbitaria bilateral sin antecedentes de traumatismo debe hacer sospechar fuertemente un neuroblastoma. Dado que se confunde fácilmente con maltrato infantil, es necesaria una evaluación sistémica que incluya ecografía abdominal y prueba de catecolaminas en orina para el diagnóstico diferencial.
Q¿Qué es el síndrome de opsoclono-mioclono?
A
Es un síndrome caracterizado por movimientos oculares sacádicos de alta frecuencia (10-15 Hz) y multidireccionales acompañados de mioclonías. Se considera un síndrome paraneoplásico causado por anticuerpos anormales contra el ARN neuronal. Se sabe que el 48% de los pacientes con OMS presentan neuroblastoma latente, por lo que la evaluación sistémica es esencial en niños con OMS.
La mayoría de los casos son esporádicos y no se han identificado factores de riesgo claros.
Factores genéticos (aproximadamente 1-2% de todos los casos):
Mutación germinal de ALK: Causa más común de NB familiar
Anomalía de PHOX2B: Asociada con alteración de la diferenciación a neuronas maduras
Anomalía de KIF1B / amplificación de MYCN: Parte del NB familiar
Variación en el número de copias de NBPF10 (deleción 1p/11q): Asociada con NB de alto riesgo
Duplicación del gen LMO1: Factor de riesgo para NB agresivo
Asociación con NF1 y síndrome de Beckwith-Wiedemann: Rara vez coexiste
Mutaciones genéticas asociadas con el NB esporádico:
Variación genética en 6p22: Se ha informado asociación con NB esporádico
Amplificación de MYCN se encuentra en el 20-25% de los NB infantiles y muestra una distribución bimodal de 3-10 veces o 100-300 veces. Es un marcador molecular fuertemente asociado con NB agresivo5).
Las características de biología molecular del NB en adultos difieren de las de los niños; la amplificación de MYCN es rara. Las mutaciones de ATRX (11%), mutaciones de ALK (hasta un 14%) y reordenamientos de TERT (23%) son características5).
Factores de riesgo perinatales (según estudios de cohortes retrospectivos):
Lactantes de baja edad: alto peso al nacer, aumento de peso excesivo de la madre, hipertensión gestacional, parto a edad avanzada
Lactantes mayores: bajo peso al nacer aumenta el riesgo
Grupo de edad avanzada: primer hijo, primera cesárea, parto prolongado, rotura prematura de membranas
Uso materno de medicamentos (diuréticos, antihipertensivos), anemia durante el embarazo, uso temporal de tintes para el cabello
No se conocen factores de riesgo específicos para la metástasis orbitaria.
Catecolaminas en orina (HVA/VMA): Anteriormente se reportaba elevación en el 90–95% de los casos, pero estudios recientes muestran una sensibilidad más baja. En adultos, solo es del 40–57% (en contraste con el 95% en niños).
Análisis de sangre: Hemograma completo, función hepática y renal, electrolitos, ferritina, LDH, función tiroidea.
Biopsia de médula ósea: Aspiración y biopsia con aguja de las crestas ilíacas posteriores bilaterales para evaluar la infiltración de la médula ósea.
Prueba de anticuerpos: El anticuerpo anti-Hu (ANNA-1) es útil para el diagnóstico del neuroblastoma infantil.
Punción lumbar: Se evita a menos que se conozca metástasis en SNC (hay informes de aumento de metástasis en SNC después de PL)
Hipointenso en T1, heterogéneo en T2, evaluación de extensión intracraneal
Examen detallado intracraneal y orbitario
MIBG (123I)
Óptimo para identificar metástasis en tejidos blandos y hueso
Evaluación de metástasis en todo el cuerpo
PET
Alta sensibilidad y especificidad
Diagnóstico y monitorización del tratamiento
La gammagrafía con MIBG (123I-MIBG) es superior a la PET/TC en la identificación de metástasis de tejidos blandos y óseas. En las metástasis orbitarias, la TC muestra cambios osteolíticos, y la RM muestra isointensidad con los músculos extraoculares e hipointensidad respecto a la grasa en T1, e hiperintensidad respecto a los músculos extraoculares y la grasa en T2. Son frecuentes las metástasis en la pared posterolateral de la órbita.
Q¿Cuál es la prueba de imagen más útil para el neuroblastoma?
A
La gammagrafía con MIBG (123I-MIBG) se considera la más útil para identificar metástasis de tejidos blandos y óseas, y es superior a la PET/TC. Debido a que puede evaluar todas las lesiones metastásicas en todo el cuerpo a la vez, se utiliza tanto para la estadificación inicial como para la evaluación de la respuesta al tratamiento.
Características de las células tumorales: Neuroblastos inmaduros (10–15 μm de diámetro). Núcleos vesiculares grandes e hipercromáticos, citoplasma eosinofílico escaso
Patrones de disposición: Disposición en láminas, nidos o cordones; formación de pseudorrosetas (tipo Homer-Wright)
La resección total rara vez está indicada para tumores orbitarios metastásicos. Se realiza un tratamiento eficaz contra el cáncer primario, como quimioterapia o radioterapia. En el neuroblastoma pediátrico, si responde a la quimioterapia, el pronóstico es relativamente bueno. La tasa de supervivencia a 5 años con metástasis orbitaria es muy baja, del 7,6%.
No existe un protocolo estándar establecido y a menudo se adaptan los protocolos pediátricos5). Se realiza cirugía (incluyendo laparoscópica) + quimioterapia (como carboplatino + etopósido). La tasa de supervivencia general a 5 años en adultos de 20 años o más es del 36,3%, lo que indica un mal pronóstico5).
Q¿Cuál es el pronóstico cuando el neuroblastoma hace metástasis en la órbita?
A
La tasa de supervivencia a 5 años del neuroblastoma con metástasis orbitaria es muy baja, del 7,6%. La metástasis orbitaria a menudo indica enfermedad metastásica a distancia (equivalente a estadio M), y se realiza un tratamiento multidisciplinario como grupo de alto riesgo, pero hay límites en la mejora del pronóstico.
El NB se origina a partir de células precursoras del sistema nervioso simpático (simpatoblastos). Las mutaciones en PHOX2B alteran la diferenciación hacia neuronas maduras, lo que conduce a la tumorigénesis. Las mutaciones en ALK se asocian con una disminución de la proliferación y un aumento de neuronas simpáticas inmaduras.
Anomalía del metabolismo de las catecolaminas: Debido a un defecto en la síntesis de catecolaminas en las células tumorales, los metabolitos intermedios HVA y VMA se acumulan y se excretan en la orina. Esto constituye la base de la prueba de catecolaminas en orina.
La activación de la vía de las poliaminas mediante la amplificación de MYCN promueve el crecimiento tumoral3).
MYCN (factor de transcripción de la familia MYC) → activación transcripcional de ODC1 → promoción de la síntesis de poliaminas (ornitina → putrescina → espermidina → espermina) → activación de eIF5A → activación de LIN28 → supresión de Let-7 miRNA → promoción del crecimiento tumoral
Síndrome de opsoclono-mioclono (OMS): Causado por anticuerpos anormales contra el ARN neuronal. Se considera un síntoma paraneoplásico por reactividad cruzada con el tejido.
Mecanismo de la discapacidad visual (sin compresión directa): La respuesta inmune al NB reacciona de forma cruzada con el tejido. También pueden estar implicados los metabolitos tóxicos derivados del cáncer y la detención del transporte axonal inducida por quimioterapia.
Algunos NB (especialmente en estadio MS) experimentan regresión espontánea. Se cree que los mecanismos implican hipermetilación del ADN subtelomérico, apoptosis, deficiencia del factor de crecimiento nervioso (NGF) y respuesta inmune.
Características moleculares biológicas del NB pediátrico vs. adulto
Los niños y los adultos tienen características genéticas muy diferentes1)5).
Característica
NB pediátrico
NB adulto
Amplificación de MYCN
20–25%
Raro
Expresión de PHOX2B
Alta (alta sensibilidad y especificidad)
50% negativo
Mutación de ATRX
Rara
11%
Mutación de ALK
Rara
Hasta 14%
Reordenamiento de TERT
Raro
23%
El hecho de que el 50% de los NB adultos sean PHOX2B negativos sugiere la posibilidad de un linaje celular diferente (como el origen tímico) en comparación con el tipo pediátrico1).
7. Investigación más reciente y perspectivas futuras (informes en fase de investigación)
El 13 de diciembre de 2023, la FDA de EE. UU. aprobó un nuevo fármaco para la terapia de mantenimiento del NB de alto riesgo3).
Jiang & Yu et al. (2024) detallaron el perfil farmacológico de la eflornitina, informando que es un inhibidor irreversible (inhibidor suicida) de la ODC (ornitina descarboxilasa), dirigido a la vía de las poliaminas en el NB con amplificación de MYCN3). Se completó un ensayo de fase 2 (NCT02395666) como terapia de mantenimiento después de la inmunoterapia (dinutuximab), lo que llevó a la aprobación de la FDA.
Propiedades del fármaco:
Peso molecular: 182.2 g/mol
Vía de administración: Oral
Vida media: Aproximadamente 3.5 horas (excreción renal, casi no se metaboliza)
Indicación: terapia de mantenimiento después de inmunoterapia para neuroblastoma de alto riesgo
Se están explorando estrategias de tratamiento personalizadas utilizando planificación terapéutica digital (por ejemplo, Oncompass™)5).
Q¿Qué es la eflornitina (IWILFIN)?
A
Es un fármaco oral aprobado por la FDA en diciembre de 2023 como terapia de mantenimiento después de la inmunoterapia para el neuroblastoma de alto riesgo. Inhibe irreversiblemente la ODC (ornitina descarboxilasa), suprimiendo la síntesis de poliaminas y bloqueando las señales de crecimiento tumoral impulsadas por la amplificación de MYCN 3). El estado de aprobación en Japón debe confirmarse con el médico tratante.
Collins K, Ulbright TM, Davis JL. Anterior mediastinal neuroblastoma in an adult: an additional case of a rare tumor in an unusual location with review of the literature. Diagn Pathol. 2023;18:127.
Hu J, Xia B, Yuan X, et al. Neuroblastoma with superficial soft tissue mass as the first symptom: case reports with atypical ultrasonic image and literature review. Braz J Med Biol Res. 2023;56:e12975.
Jiang J, Yu Y. Eflornithine for treatment of high-risk neuroblastoma. Trends Pharmacol Sci. 2024;45(6):577-578.
do Amaral-Silva GK, Leite AA, Mariz BALA, et al. Metastatic neuroblastoma to the mandible of children: report of two cases and critical review of the literature. Head Neck Pathol. 2021;15:757-768.
Telecan T, Andras I, Bungardean MR, et al. Adrenal gland primary neuroblastoma in an adult patient: a case report and literature review. Medicina. 2023;59:33.
Copia el texto del artículo y pégalo en el asistente de IA que prefieras.
Artículo copiado al portapapeles
Abre un asistente de IA abajo y pega el texto copiado en el chat.