第1期
中心凹旁RPE改变:视网膜色素上皮(RPE)出现点状或斑状色素变化。
视力及自觉症状:多保持正常。

中心性轮纹状脉络膜营养不良(Central Areolar Choroidal Dystrophy; CACD)是一种遗传性黄斑营养不良,表现为黄斑部边界清晰的视网膜脉络膜萎缩。发病率低(约10万人中1人),属于罕见病。
发病多在20~50岁。遗传方式以常染色体显性遗传(AD)为主,也有常染色体隐性遗传(AR)和散发病例的报道。
最常见的致病基因是PRPH2(染色体17p13),编码peripherin-2蛋白。Peripherin-2对感光细胞外节盘膜的形成和稳定至关重要。单倍剂量不足(一个等位基因功能丧失)被认为是主要发病机制。
CACD与ABCA4基因突变引起的疾病(如Stargardt病)在表型上有相似之处1),基因检测对鉴别诊断很重要。未见全身并发症的报道。
主要症状是双眼中心暗点(中心部视物不清)。视力下降有时早期即可出现,但也可能直到病程进展仍保持较好视力。
双眼对称性黄斑病变,病期分为4期。视神经、视网膜血管及周边视网膜保持正常。萎缩区域大小约为视盘直径(DD)的2~4倍。
第1期
中心凹旁RPE改变:视网膜色素上皮(RPE)出现点状或斑状色素变化。
视力及自觉症状:多保持正常。
第2期
边界不清的低色素萎缩:中心凹外出现淡色萎缩区域。
RPE变化扩大:可见边界不清的萎缩。
第3期
边界清晰的RPE萎缩:中心凹外形成边界清晰的萎缩区域。
中心凹保留:此阶段中心凹(fovea)尚存,视力相对保持。
第4期
累及中心凹的完全萎缩:萎缩区域扩展至包括中心凹在内的整个黄斑。
严重视力障碍:脉络膜毛细血管、感光细胞和RPE广泛丧失,导致视力显著下降。
进展速度个体差异很大。通常病程缓慢,有些病例可能需要数十年才进入第4期。但基因型不同进展速度可能不同,定期眼科评估至关重要。详细内容见“诊断与检查方法”一节中描述的mfERG,可作为早期进展的指标。
CACD的主要原因是PRPH2基因突变。PRPH2编码peripherin-2,参与感光细胞外节盘膜的形成和稳定。突变均妨碍感光细胞正常外节结构的维持。
由于是常染色体显性遗传(AD),携带突变基因的父母遗传给子女的概率为50%。如有近亲发病史,建议进行遗传咨询。
目前尚未明确确定后天风险因素或生活方式可促进发病或进展。
CACD主要为常染色体显性遗传,患者子女的遗传风险高达50%。通过基因检测识别突变可实现确诊和家族筛查。此外,记录突变很重要,因为将来可能成为基因治疗的对象。
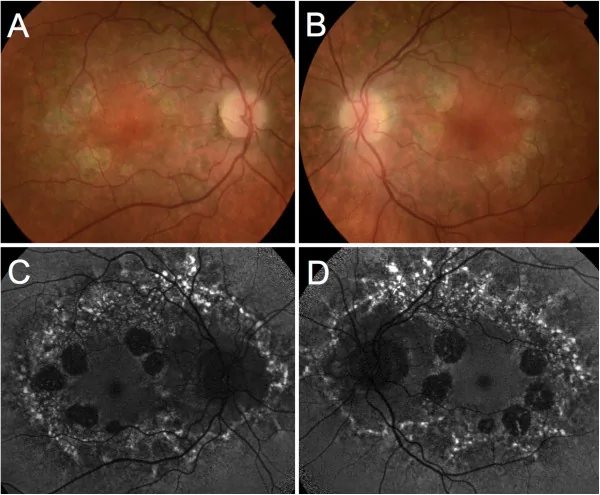
使用裂隙灯显微镜和眼底镜观察黄斑部边界清晰的萎缩区域。评估脉络膜血管暴露和色素沉着的存在。
根据病期不同,各检查所见也不同。下表显示各病期的特征性所见。
| 检查 | 早期所见 | 进展期所见 |
|---|---|---|
| FAF(眼底自发荧光) | 荧光增强 | 荧光消失(萎缩区域) |
| FA(荧光造影) | 旁中心凹高荧光 | 萎缩区域的窗样缺损 |
| OCT | POS-RPE增厚 | 外层(感光细胞和RPE)消失 |
与呈现类似黄斑萎缩的疾病进行鉴别非常重要。
| 鉴别疾病 | 鉴别要点 |
|---|---|
| 萎缩型AMD | 玻璃膜疣、边界不规则、老年发病 |
| Stargardt病 | 暗脉络膜、ABCA4突变 |
| 锥体营养不良 | 视网膜电图锥体反应显著降低、畏光为主要症状 |
目前,CACD尚无确定的治疗方法。治疗目标是控制症状和维持生活质量。
低视力康复
放大镜和遮光眼镜:开具辅助器具,以最大限度地利用剩余视功能。
职业和生活训练:支持学习适应视力障碍的日常生活技能。
遗传咨询
向家属提供信息:解释常染色体显性遗传(50%的遗传风险)。
基因检测:识别突变对于确诊和未来治疗选择很重要。
基因治疗(研究阶段)
未来的治疗候选:针对PRPH2突变的基因替代疗法研究正在进行中。
现状:目前尚不能作为常规医疗提供。
在萎缩区域,脉络膜毛细血管、RPE和感光细胞(视杆细胞和视锥细胞)消失。外核层(ONL)细胞数量显著减少,外界膜(OLM)直接与Bruch膜接触。中大型脉络膜血管即使在疾病晚期也能长期保留。
Peripherin-2定位于感光细胞外节盘边缘(rim区域),在盘的形成、稳定和维持边缘弯曲中起关键作用。
PRPH2单倍体不足(一个功能拷贝缺失)会损害正常外节形态的形成。外节结构破坏导致感光细胞凋亡,进而引起RPE和脉络膜毛细血管的继发性萎缩。
PRPH2在视杆细胞和视锥细胞中可能具有不同功能。在PRPH2基因敲除小鼠模型中,蓝色视锥细胞的变性速度比其他视锥细胞更慢。这种视杆与视锥之间的差异被认为与疾病临床表型的多样性有关。
多焦视网膜电图(mfERG)可在眼底检查可见的萎缩区域之外的旁中心凹区域检测到功能下降。这表明CACD病变范围比临床可见的萎缩更广泛。
针对包括PRPH2突变在内的遗传性视网膜疾病的基因补充疗法和基因编辑疗法的研究正在全球范围内进行。对于相关疾病(无脉络膜症),2014年在英国进行了首次基因治疗临床试验。
随着基因组医学的发展,每种突变的致病性分析正在变得更加精确。全外显子组测序(WES)的普及大大提高了诊断准确性1),使得更多患者的突变得以识别。
PRPH2在视杆细胞和视锥细胞中具有不同功能作用的分子机制正在被阐明。这一知识对于解释表型的多样性(视杆细胞主导还是视锥细胞主导的变性)很重要,并可能影响未来治疗靶点的选择。