Stage 1
Parafoveal RPE changes: Dotty or patchy pigment changes appear in the retinal pigment epithelium (RPE).
Visual acuity and symptoms: Often remains nearly normal.

Central Areolar Choroidal Dystrophy (CACD) is an inherited macular dystrophy that causes well-demarcated chorioretinal atrophy in the macula. It is rare (about 1 in 100,000 people) and classified as a rare disease.
Onset is most common between the ages of 20 and 50. The inheritance pattern is mainly autosomal dominant (AD), but autosomal recessive (AR) and sporadic cases have also been reported.
The most frequent causative gene is PRPH2 (chromosome 17p13), which encodes the peripherin-2 protein. Peripherin-2 is essential for the formation and stabilization of photoreceptor outer segment discs. Haploinsufficiency (loss of function of one allele) is thought to be the main mechanism of disease.
CACD has been noted to have phenotypic similarities with diseases caused by ABCA4 gene mutations (such as Stargardt disease) 1), and genetic testing is important for differential diagnosis. No systemic complications have been reported.
Age-related macular degeneration usually occurs after age 60 and often involves drusen or exudative changes. CACD typically occurs in younger to middle-aged adults, is characterized by well-demarcated atrophic lesions, and does not involve drusen. If a PRPH2 mutation is confirmed by genetic testing, a diagnosis of CACD can be made.
The main symptom is bilateral central scotoma (difficulty seeing in the center). Visual acuity may decrease early in some cases, but relatively good vision may be maintained until the disease progresses.
Both eyes show symmetric macular lesions, and the disease is classified into four stages. The optic nerve, retinal vessels, and peripheral retina are preserved. The size of the atrophic area is generally about 2 to 4 times the optic disc diameter (DD).
Stage 1
Parafoveal RPE changes: Dotty or patchy pigment changes appear in the retinal pigment epithelium (RPE).
Visual acuity and symptoms: Often remains nearly normal.
Stage 2
Ill-defined hypopigmented atrophy: Pale atrophic areas appear outside the fovea.
Expansion of RPE changes: Atrophy with unclear borders is observed.
Stage 3
Well-defined RPE atrophy: Clearly demarcated atrophic areas form outside the fovea.
Foveal sparing: At this stage, the fovea remains intact, and visual acuity is relatively preserved.
Stage 4
Complete atrophy involving the fovea: The atrophic area expands to include the entire macula including the fovea.
Severe visual impairment: Widespread loss of the choriocapillaris, photoreceptors, and RPE leads to a marked decrease in visual acuity.
The rate of progression varies greatly among individuals. Generally, it follows a slow course, and some cases may take decades to reach stage 4. However, the progression rate may differ depending on the genotype, and regular ophthalmic evaluation is essential. The mfERG, described in detail in the “Diagnosis and Testing Methods” section, serves as an indicator of early progression.
The main cause of CACD is mutations in the PRPH2 gene. PRPH2 encodes peripherin-2, which functions in the formation and stabilization of photoreceptor outer segment discs. Mutations prevent the maintenance of normal outer segment structure in photoreceptors.
Because it is an autosomal dominant (AD) disorder, the probability of inheriting the mutated gene from an affected parent to a child is 50%. If there is a family history of the disease in close relatives, genetic counseling is recommended.
Acquired risk factors or lifestyle factors that promote onset or progression have not been clearly identified at this time.
CACD is primarily autosomal dominant, and the genetic risk to a patient’s children is 50%. Identifying the mutation through genetic testing enables definitive diagnosis and family screening. Additionally, recording the mutation is important because it may become a target for future gene therapy.
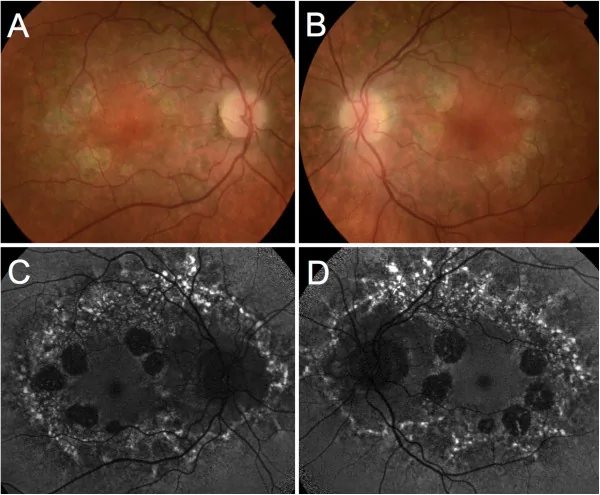
Using a slit-lamp microscope and ophthalmoscope, a well-defined atrophic area in the macula is observed. The presence of exposed choroidal vessels and pigmentation is evaluated.
Findings vary depending on the disease stage. The table below shows characteristic findings by stage.
| Examination | Early findings | Advanced findings |
|---|---|---|
| FAF (fundus autofluorescence) | Hyperautofluorescence | Loss of autofluorescence (atrophic area) |
| FA (fluorescein angiography) | Parafoveal hyperfluorescence | Window defect in atrophic area |
| OCT | POS-RPE thickening | Loss of outer layers (photoreceptors and RPE) |
It is important to differentiate from diseases that present with similar macular atrophy.
| Differential Diagnosis | Key Differentiating Features |
|---|---|
| Atrophic AMD | Drusen, irregular borders, late onset |
| Stargardt disease | Dark choroid, ABCA4 mutation |
| Cone dystrophy | Markedly reduced cone ERG responses, photophobia as main symptom |
Currently, there is no established treatment for CACD. Treatment aims to manage symptoms and maintain quality of life.
Low Vision Rehabilitation
Magnifiers and tinted glasses: Prescribe assistive devices to maximize remaining visual function.
Occupational and daily living training: Support the acquisition of daily living skills adapted to visual impairment.
Genetic Counseling
Information for families: Explain autosomal dominant inheritance (50% genetic risk).
Genetic testing: Identifying mutations is important for definitive diagnosis and future treatment options.
Gene Therapy (Research Stage)
Future treatment candidate: Research on gene replacement therapy targeting PRPH2 mutations is ongoing.
Current status: Not yet available as standard medical care.
Research on gene therapy for inherited retinal diseases including PRPH2 is accelerating worldwide. In the UK, a gene therapy trial for choroideremia was conducted in 2014, and its application to PRPH2 mutation diseases is also under study. However, it is not yet established as a standard treatment and remains at the research stage.
In the atrophic area, the choriocapillaris, RPE, and photoreceptors (rods and cones) disappear. The number of cells in the outer nuclear layer (ONL) is markedly reduced, and the outer limiting membrane (OLM) comes into direct contact with Bruch’s membrane. Medium and large choroidal vessels are preserved for a long time even in advanced stages.
Peripherin-2 is localized at the rim region of photoreceptor outer segment discs and plays an essential role in disc formation, stabilization, and maintaining rim curvature.
PRPH2 haploinsufficiency (loss of one functional copy) impairs normal outer segment morphogenesis. Disruption of outer segment structure leads to photoreceptor apoptosis, followed by secondary atrophy of the RPE and choriocapillaris.
PRPH2 is suggested to have different functions in rods and cones. In PRPH2 knockout mouse models, blue cones degenerate more slowly than other cones. This difference between rods and cones is thought to contribute to the diversity of clinical phenotypes.
Multifocal electroretinography (mfERG) detects functional decline in the parafoveal area beyond the atrophic region visible on fundus examination. This indicates that CACD lesions extend more widely than the clinically visible atrophy.
Research on gene replacement therapy and gene editing therapy for inherited retinal diseases including PRPH2 mutations is being conducted worldwide. For a related disease (choroideremia), the first gene therapy clinical trial was performed in the UK in 2014.
Pathogenicity analysis of each mutation is becoming more precise with the progress of genomic medicine. The widespread use of whole exome sequencing (WES) has greatly improved diagnostic accuracy 1), enabling identification of mutations in more patients.
It is being elucidated at the molecular level that PRPH2 has different functional roles in rods and cones. This knowledge is important for explaining the diversity of phenotypes (rod-dominant vs. cone-dominant degeneration) and may influence the selection of future therapeutic targets.