ระยะที่ 1
การเปลี่ยนแปลงของ RPE รอบรอยบุ๋มจอตา: การเปลี่ยนแปลงของเม็ดสีแบบจุดหรือปื้นในเยื่อบุผิวเม็ดสีจอตา (RPE)
การมองเห็นและอาการที่ผู้ป่วยรับรู้: มักเป็นปกติ

โรคจอประสาทตาเสื่อมชนิดวงแหวนกลาง (Central Areolar Choroidal Dystrophy; CACD) เป็นโรคจอประสาทตาเสื่อมที่ถ่ายทอดทางพันธุกรรม ทำให้เกิดการฝ่อของจอตาและคอรอยด์อย่างชัดเจนบริเวณจุดรับภาพ อุบัติการณ์ต่ำ (ประมาณ 1 ใน 100,000 คน) และจัดเป็นโรคหายาก
โรคมักเริ่มในช่วงอายุ 20–50 ปี รูปแบบการถ่ายทอดทางพันธุกรรมส่วนใหญ่เป็นแบบ autosomal dominant (AD) แต่ก็มีรายงานแบบ autosomal recessive (AR) และแบบประปราย
ยีนที่พบบ่อยที่สุดคือ PRPH2 (โครโมโซม 17p13) ซึ่งสร้างโปรตีน peripherin-2 โปรตีนนี้จำเป็นต่อการสร้างและคงสภาพของแผ่นจานส่วนนอกของเซลล์รับแสง การขาด haploinsufficiency (การสูญเสียการทำงานของอัลลีลหนึ่ง) ถือเป็นกลไกหลัก
CACD มีลักษณะฟีโนไทป์คล้ายกับโรคที่เกิดจากการกลายพันธุ์ของยีน ABCA4 (เช่น โรค Stargardt) 1) ดังนั้นการตรวจยีนจึงสำคัญในการวินิจฉัยแยกโรค ไม่มีรายงานภาวะแทรกซ้อนทั่วร่างกาย
AMD มักเริ่มหลังอายุ 60 ปี และมักมี drusen และการเปลี่ยนแปลงแบบ exudative CACD เริ่มตั้งแต่อายุน้อยถึงวัยกลางคน มีการฝ่อชัดเจนโดยไม่มี drusen หากตรวจพบการกลายพันธุ์ของ PRPH2 จากการตรวจยีน ก็สามารถวินิจฉัย CACD ได้
อาการหลักคือจุดบอดกลางตาทั้งสองข้าง (มองเห็นส่วนกลางได้ยาก) การมองเห็นลดลงอาจเกิดขึ้นเร็ว แต่ในบางกรณีการมองเห็นยังค่อนข้างดีจนถึงระยะท้าย
รอยโรคที่จอประสาทตาสมมาตรทั้งสองข้าง แบ่งระยะเป็น 4 ระยะ เส้นประสาทตา หลอดเลือดจอประสาทตา และจอประสาทตาส่วนปลายยังคงปกติ ขนาดของบริเวณฝ่อประมาณ 2-4 เท่าของเส้นผ่านศูนย์กลางของหัวประสาทตา (DD)
ระยะที่ 1
การเปลี่ยนแปลงของ RPE รอบรอยบุ๋มจอตา: การเปลี่ยนแปลงของเม็ดสีแบบจุดหรือปื้นในเยื่อบุผิวเม็ดสีจอตา (RPE)
การมองเห็นและอาการที่ผู้ป่วยรับรู้: มักเป็นปกติ
ระยะที่ 2
รอยฝ่อสีจางขอบเขตไม่ชัดเจน: บริเวณฝ่อสีซีดปรากฏขึ้นนอกรอยบุ๋มจอตา
การขยายตัวของการเปลี่ยนแปลง RPE: พบรอยฝ่อที่ขอบเขตยังไม่ชัดเจน
ระยะที่ 3
รอยฝ่อของ RPE ขอบเขตชัดเจน: บริเวณฝ่อที่มีขอบเขตชัดเจนก่อตัวขึ้นนอกรอยบุ๋มจอตา
การคงไว้ของรอยบุ๋มจอตา: ในระยะนี้ รอยบุ๋มจอตา (fovea) ยังคงอยู่ การมองเห็นค่อนข้างคงที่
ระยะที่ 4
ฝ่อสมบูรณ์ที่รวมรอยบุ๋มจอตา (fovea): บริเวณฝ่อขยายครอบคลุมรอยบุ๋มจอตาและจอประสาทตาทั้งหมด
ความบกพร่องทางการมองเห็นอย่างรุนแรง: การสูญเสียหลอดเลือดฝอยคอรอยด์ เซลล์รับแสง และ RPE อย่างกว้างขวางทำให้การมองเห็นลดลงอย่างมาก
ความเร็วในการดำเนินโรคแตกต่างกันมากในแต่ละบุคคล โดยทั่วไปแล้วจะเป็นไปอย่างช้าๆ และบางรายอาจใช้เวลาหลายสิบปีกว่าจะถึงระยะที่ 4 อย่างไรก็ตาม อัตราการดำเนินโรคอาจแตกต่างกันตามจีโนไทป์ และการประเมินทางจักษุวิทยาเป็นระยะเป็นสิ่งจำเป็น mfERG ซึ่งอธิบายโดยละเอียดในหัวข้อ «การวินิจฉัยและวิธีการตรวจ» เป็นตัวบ่งชี้การดำเนินโรคในระยะแรก
สาเหตุหลักของ CACD คือการกลายพันธุ์ในยีน PRPH2 ยีน PRPH2 เข้ารหัสโปรตีนเพอริเฟอริน-2 ซึ่งทำหน้าที่ในการสร้างและทำให้แผ่นดิสก์ของส่วนนอกเซลล์รับแสงคงที่ การกลายพันธุ์ขัดขวางการรักษาโครงสร้างส่วนนอกของเซลล์รับแสงตามปกติ
เนื่องจากเป็นพันธุกรรมแบบออโตโซมอลโดมิแนนต์ (AD) ความน่าจะเป็นในการถ่ายทอดยีนกลายพันธุ์จากพ่อแม่สู่ลูกคือ 50% หากมีประวัติครอบครัว แนะนำให้รับคำปรึกษาทางพันธุกรรม
ปัจจัยเสี่ยงที่ได้มาหรือปัจจัยการดำเนินชีวิตที่เร่งการเริ่มต้นหรือการดำเนินโรคยังไม่ถูกระบุอย่างชัดเจนในปัจจุบัน
CACD ส่วนใหญ่เป็นพันธุกรรมแบบออโตโซมอลโดมิแนนต์ และความเสี่ยงในการถ่ายทอดสู่บุตรของผู้ป่วยสูงถึง 50% การกลายพันธุ์สามารถระบุได้โดยการตรวจทางพันธุกรรม ทำให้สามารถวินิจฉัยที่แน่นอนและคัดกรองครอบครัวได้ นอกจากนี้ เนื่องจากอาจเป็นเป้าหมายของการบำบัดด้วยยีนในอนาคต การบันทึกการกลายพันธุ์จึงมีความสำคัญ
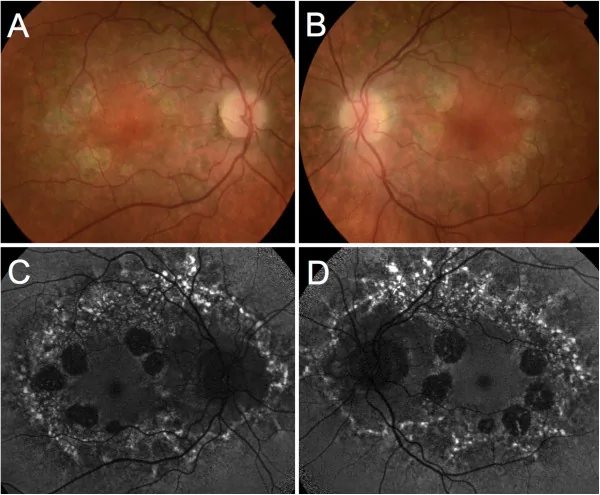
ด้วยกล้องจุลทรรศน์ชนิดกรีด (slit-lamp) และจักษุแพทย์ (ophthalmoscope) ยืนยันบริเวณฝ่อที่ชัดเจนในจอประสาทตาส่วนกลาง (macula) ประเมินการเปิดเผยของหลอดเลือดคอรอยด์หรือการมีเม็ดสี
ผลการตรวจแต่ละอย่างแตกต่างกันไปตามระยะของโรค ตารางด้านล่างแสดงผลการตรวจที่มีลักษณะเฉพาะตามระยะ
| การตรวจ | ผลการตรวจระยะแรก | ผลการตรวจระยะลุกลาม |
|---|---|---|
| FAF (การเรืองแสงอัตโนมัติของจอตา) | การเรืองแสงมากเกินไป | การสูญเสียการเรืองแสง (บริเวณฝ่อ) |
| FA (การถ่ายภาพหลอดเลือดด้วยฟลูออเรสซีน) | การเรืองแสงมากเกินไปรอบรอยบุ๋มจอตา | การเติมเต็มแบบโปร่งแสงในบริเวณฝ่อ |
| OCT | ความหนาของ POS-RPE เพิ่มขึ้น | การสูญเสียชั้นนอก (เซลล์รับแสงและ RPE) |
สิ่งสำคัญคือต้องแยกความแตกต่างจากโรคที่มีจอประสาทตาฝ่อคล้ายกัน
| โรคที่ต้องแยก | จุดที่ใช้แยก |
|---|---|
| จอประสาทตาเสื่อมตามอายุชนิดฝ่อ (AMD ชนิดฝ่อ) | ดรูเซน ขอบเขตไม่สม่ำเสมอ เกิดในวัยสูงอายุ |
| โรคสตาร์การ์ด | คอรอยด์มืด การกลายพันธุ์ของ ABCA4 |
| โรคเซลล์รูปกรวยเสื่อม | การตอบสนองของเซลล์รูปกรวยใน ERG ลดลงอย่างมาก อาการหลักคือกลัวแสง |
ปัจจุบันยังไม่มีการรักษาที่แน่ชัดสำหรับ CACD การรักษามุ่งเน้นการจัดการอาการและรักษาคุณภาพชีวิต
การฟื้นฟูสมรรถภาพสำหรับผู้มีสายตาเลือนราง
แว่นขยายและแว่นตากันแสง: จ่ายอุปกรณ์ช่วยเหลือเพื่อใช้ประโยชน์สูงสุดจากการมองเห็นที่เหลืออยู่
การฝึกอาชีพและการใช้ชีวิต: ช่วยเหลือในการเรียนรู้ทักษะชีวิตประจำวันที่ปรับให้เข้ากับความบกพร่องทางการมองเห็น
การให้คำปรึกษาทางพันธุกรรม
การให้ข้อมูลแก่ครอบครัว: อธิบายเกี่ยวกับการถ่ายทอดทางพันธุกรรมแบบออโตโซมอลโดมิแนนต์ (ความเสี่ยงในการถ่ายทอด 50%)
การตรวจทางพันธุกรรม: การระบุการกลายพันธุ์มีความสำคัญต่อการวินิจฉัยที่แน่นอนและทางเลือกการรักษาในอนาคต
การบำบัดด้วยยีน (ระยะวิจัย)
ตัวเลือกการรักษาในอนาคต: กำลังมีการวิจัยเกี่ยวกับการบำบัดด้วยการแทนที่ยีนที่มุ่งเป้าไปที่การกลายพันธุ์ของ PRPH2
สถานะปัจจุบัน: ยังไม่สามารถให้บริการเป็นการรักษาทั่วไปได้ในขณะนี้
การวิจัยเกี่ยวกับการบำบัดด้วยยีนสำหรับโรคจอประสาทตาทางพันธุกรรมที่เกี่ยวข้องกับ PRPH2 กำลังเร่งตัวขึ้นทั่วโลก ในสหราชอาณาจักร มีการทดลองการบำบัดด้วยยีนสำหรับโรคคอรอยเดอเรเมียในปี 2014 และกำลังศึกษาการประยุกต์ใช้กับโรคที่เกิดจากการกลายพันธุ์ของ PRPH2 อย่างไรก็ตาม ในปัจจุบันยังไม่ได้รับการยอมรับว่าเป็นการรักษามาตรฐานและยังอยู่ในขั้นตอนการวิจัย
ในบริเวณที่ฝ่อ เส้นเลือดฝอยคอรอยด์ เซลล์เยื่อบุผิวรงควัตถุจอประสาทตา และเซลล์รับแสง (แท่งและกรวย) จะหายไป จำนวนเซลล์ในชั้นนิวเคลียสชั้นนอกลดลงอย่างมาก และเยื่อลิมิตันส์ชั้นนอกสัมผัสโดยตรงกับเยื่อบรูค หลอดเลือดคอรอยด์ขนาดกลางและใหญ่ยังคงอยู่เป็นเวลานานแม้ในระยะลุกลาม
โปรตีนเพอริเฟอริน-2 อยู่บริเวณขอบของแผ่นดิสก์ส่วนนอกของเซลล์รับแสง และมีบทบาทสำคัญในการสร้างแผ่นดิสก์ การทำให้คงตัว และการรักษาความโค้งของขอบ
เมื่อเกิดภาวะ PRPH2 แฮพลอยด์ไม่เพียงพอ (สูญเสียการทำงานของหนึ่งสำเนา) การสร้างสัณฐานวิทยาของส่วนนอกตามปกติจะบกพร่อง การพังทลายของโครงสร้างส่วนนอกจะดำเนินไปสู่การตายแบบอะพอพโทซิสของเซลล์รับแสง ซึ่งนำไปสู่การฝ่อทุติยภูมิของเซลล์เยื่อบุผิวรงควัตถุจอประสาทตาและเส้นเลือดฝอยคอรอยด์
มีข้อเสนอแนะว่า PRPH2 ทำหน้าที่แตกต่างกันในเซลล์แท่งและเซลล์กรวย ในหนูที่ถูกน็อกเอาต์ PRPH2 มีรายงานว่าเซลล์กรวยสีน้ำเงินเสื่อมช้ากว่าเซลล์กรวยอื่นๆ ความแตกต่างระหว่างเซลล์แท่งและเซลล์กรวยนี้เชื่อว่ามีส่วนทำให้เกิดความหลากหลายของฟีโนไทป์ทางคลินิกของโรค
ในการตรวจคลื่นไฟฟ้าจอประสาทตาแบบหลายจุด จะตรวจพบการทำงานที่ลดลงแม้ในบริเวณพาราฟอฟเวียที่เกินกว่าบริเวณฝ่อที่มองเห็นได้จากการตรวจอวัยวะรับภาพ ซึ่งบ่งชี้ว่ารอยโรค CACD แผ่ขยายกว้างกว่าบริเวณฝ่อที่มองเห็นได้ทางคลินิก
การวิจัยเกี่ยวกับการบำบัดด้วยการทดแทนยีนและการบำบัดด้วยการตัดต่อยีนสำหรับโรคจอประสาทตาทางพันธุกรรมที่เกี่ยวข้องกับการกลายพันธุ์ของ PRPH2 กำลังดำเนินการในระดับโลก สำหรับโรคที่เกี่ยวข้อง (choroidermia) การทดลองทางคลินิกการบำบัดด้วยยีนครั้งแรกดำเนินการในสหราชอาณาจักรในปี 2014
การวิเคราะห์พยาธิสภาพของการกลายพันธุ์แต่ละชนิดได้รับการปรับปรุงให้ละเอียดขึ้นพร้อมกับความก้าวหน้าของการแพทย์จีโนม การแพร่หลายของการหาลำดับเอ็กโซมทั้งหมด (WES) ช่วยเพิ่มความแม่นยำในการวินิจฉัยอย่างมีนัยสำคัญ 1) ทำให้สามารถระบุการกลายพันธุ์ในผู้ป่วยได้มากขึ้น
PRPH2 มีบทบาทหน้าที่แตกต่างกันในเซลล์รูปแท่งและเซลล์รูปกรวยในระดับโมเลกุล ความรู้นี้มีความสำคัญในการอธิบายความหลากหลายของฟีโนไทป์ (ว่าการเสื่อมนั้นเด่นในเซลล์รูปแท่งหรือเซลล์รูปกรวย) และอาจส่งผลต่อการเลือกเป้าหมายการรักษาในอนาคต