Stadium 1
Parafoveoläre RPE-Veränderungen : punktförmige oder fleckige Pigmentveränderungen des retinalen Pigmentepithels (RPE) treten auf.
Sehschärfe und subjektive Symptome : meist normal und stabil.

Die zentrale areoläre Aderhautdystrophie (Central Areolar Choroidal Dystrophy; CACD) ist eine erbliche Makuladystrophie, die zu einer scharf begrenzten Aderhaut- und Netzhautatrophie in der Makula führt. Die Häufigkeit ist gering (etwa 1 pro 100.000 Personen) und sie wird zu den seltenen Erkrankungen gezählt.
Der Beginn liegt meist zwischen dem 20. und 50. Lebensjahr. Der Erbgang ist überwiegend autosomal-dominant (AD), aber auch autosomal-rezessive (AR) und sporadische Fälle wurden berichtet.
Das häufigste ursächliche Gen ist PRPH2 (Chromosom 17p13), das für das Protein Peripherin-2 kodiert. Peripherin-2 ist für die Bildung und Stabilität der Außensegment-Discs der Photorezeptoren unerlässlich. Haploinsuffizienz (Funktionsverlust eines Allels) wird als Hauptmechanismus angesehen.
Die CACD zeigt phänotypische Ähnlichkeiten mit Erkrankungen, die durch ABCA4-Genmutationen verursacht werden (wie Morbus Stargardt)1), und Gentests sind für die Differenzialdiagnose wichtig. Systemische Komplikationen wurden nicht berichtet.
Die AMD tritt in der Regel nach dem 60. Lebensjahr auf und geht häufig mit Drusen oder exsudativen Veränderungen einher. Die CACD tritt häufiger im jungen bis mittleren Erwachsenenalter auf, ist durch scharf begrenzte atrophische Läsionen gekennzeichnet und weist keine Drusen auf. Der Nachweis einer PRPH2-Mutation im Gentest bestätigt die Diagnose einer CACD.
Das Hauptsymptom ist ein beidseitiges zentrales Skotom (zentrale Sehstörung). Eine Sehverschlechterung kann früh auftreten, aber manchmal bleibt bis zu einem fortgeschrittenen Stadium eine relativ gute Sehschärfe erhalten.
Beide Augen zeigen symmetrische Makulaläsionen, die in 4 Stadien eingeteilt werden. Sehnerv, Netzhautgefäße und periphere Netzhaut bleiben erhalten. Die Größe der atrophischen Zone beträgt in der Regel etwa das 2- bis 4-fache des Papillendurchmessers (DD).
Stadium 1
Parafoveoläre RPE-Veränderungen : punktförmige oder fleckige Pigmentveränderungen des retinalen Pigmentepithels (RPE) treten auf.
Sehschärfe und subjektive Symptome : meist normal und stabil.
Stadium 2
Unscharf begrenzte hypopigmentierte Atrophie : eine blasse atrophische Zone erscheint außerhalb der Fovea.
Ausdehnung der RPE-Veränderungen : eine noch unscharf begrenzte Atrophie wird beobachtet.
Stadium 3
Scharf begrenzte RPE-Atrophie : eine gut abgegrenzte atrophische Zone bildet sich außerhalb der Fovea.
Fovea-Erhalt : in diesem Stadium ist die Fovea noch intakt und die Sehschärfe relativ erhalten.
Stadium 4
Vollständige Atrophie einschließlich der Fovea: Der Atrophiebereich erstreckt sich über die gesamte Makula einschließlich der Fovea.
Schwere Sehbehinderung: Durch den ausgedehnten Verlust der Choriokapillaris, der Photorezeptoren und des RPE kommt es zu einer deutlichen Verschlechterung der Sehschärfe.
Die Fortschrittsgeschwindigkeit variiert stark von Person zu Person. Im Allgemeinen verläuft sie langsam, und in manchen Fällen kann es Jahrzehnte dauern, bis Stadium 4 erreicht wird. Allerdings kann die Fortschrittsgeschwindigkeit je nach Genotyp unterschiedlich sein, und regelmäßige augenärztliche Untersuchungen sind unerlässlich. Das mfERG, das im Abschnitt „Diagnose und Untersuchungsmethoden“ ausführlich beschrieben wird, ist ein Indikator für das frühe Fortschreiten.
Die Hauptursache der CACD ist eine Mutation im PRPH2-Gen. PRPH2 kodiert für Peripherin-2, das an der Bildung und Stabilisierung der Außensegment-Discs der Photorezeptoren beteiligt ist. Mutationen beeinträchtigen die Aufrechterhaltung der normalen Außensegmentstruktur der Photorezeptoren.
Da es sich um einen autosomal-dominanten (AD) Erbgang handelt, beträgt die Wahrscheinlichkeit, dass das mutierte Gen von einem Elternteil an ein Kind weitergegeben wird, 50 %. Bei einer familiären Vorgeschichte der Erkrankung wird eine genetische Beratung empfohlen.
Erworbene Risikofaktoren oder Lebensstilfaktoren, die das Auftreten oder Fortschreiten der Erkrankung fördern, wurden derzeit nicht eindeutig identifiziert.
Die CACD wird hauptsächlich autosomal-dominant vererbt, und das genetische Risiko für die Kinder eines Patienten beträgt 50 %. Durch die Identifizierung der Mutation mittels Gentest können eine definitive Diagnose und ein Familienscreening durchgeführt werden. Da zudem eine zukünftige Gentherapie in Frage kommen könnte, ist die Dokumentation der Mutation wichtig.
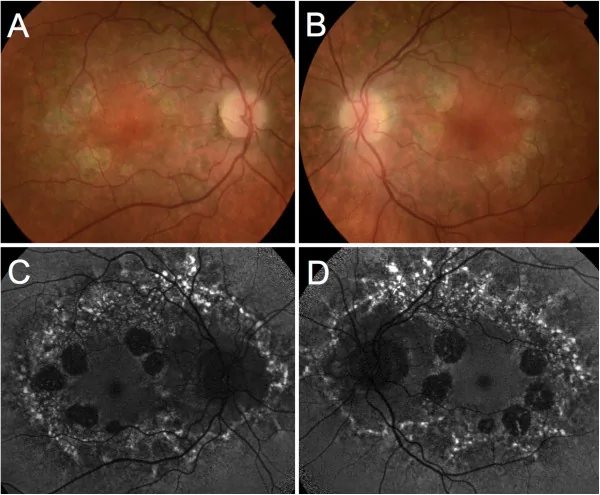
Mit Spaltlampe und Ophthalmoskop wird ein scharf begrenzter atrophischer Bereich in der Makula bestätigt. Beurteilung der Freilegung von Aderhautgefäßen und des Vorhandenseins von Pigmentablagerungen.
Die Befunde der einzelnen Untersuchungen variieren je nach Krankheitsstadium. Die folgende Tabelle zeigt die charakteristischen Befunde nach Stadium.
| Untersuchung | Frühbefunde | Fortgeschrittene Befunde |
|---|---|---|
| FAF (Fundusautofluoreszenz) | Hyperfluoreszenz | Fluoreszenzverlust (atrophischer Bereich) |
| FA (Fluoreszenzangiographie) | Parafoveale Hyperfluoreszenz | Durchscheinende Füllung des atrophischen Bereichs |
| OCT | POS-RPE-Verdickung | Verlust der äußeren Schichten (Photorezeptoren, RPE) |
Die Abgrenzung zu Erkrankungen mit ähnlicher Makulaatrophie ist wichtig.
| Differenzialdiagnose | Abgrenzungsmerkmale |
|---|---|
| Atrophische AMD | Drusen, unregelmäßige Grenzen, spätes Auftreten |
| Morbus Stargardt | Dunkle Aderhaut, ABCA4-Mutation |
| Zapfendystrophie | Stark reduzierte Zapfenantwort im ERG, Photophobie als Hauptsymptom |
Derzeit gibt es keine etablierte Behandlung für CACD. Die Behandlung zielt auf die Symptomkontrolle und die Aufrechterhaltung der Lebensqualität ab.
Sehbehindertenrehabilitation
Vergrößerungsgläser und Blendschutzbrillen : Verschreibung von Hilfsmitteln zur bestmöglichen Nutzung der verbleibenden Sehfunktion.
Berufs- und Lebenstraining : Unterstützung beim Erlernen von Alltagstechniken, die an die Sehbehinderung angepasst sind.
Genetische Beratung
Information für Familien : Erläuterung des autosomal-dominanten Erbgangs (50 % Vererbungsrisiko).
Gentests : Die Identifizierung von Mutationen ist wichtig für die endgültige Diagnose und zukünftige Behandlungsoptionen.
Gentherapie (Forschungsstadium)
Zukünftiger Behandlungskandidat : Die Forschung zur Genersatztherapie, die auf PRPH2-Mutationen abzielt, schreitet voran.
Aktueller Stand : Derzeit ist sie noch nicht als allgemeine medizinische Behandlung verfügbar.
Die Forschung zur Gentherapie für erbliche Netzhauterkrankungen, einschließlich solcher mit PRPH2, beschleunigt sich weltweit. Im Vereinigten Königreich wurde 2014 ein Gentherapieversuch für Choroiderämie durchgeführt, und die Anwendung auf PRPH2-Mutationserkrankungen wird ebenfalls untersucht. Derzeit ist sie jedoch noch nicht als Standardbehandlung etabliert und befindet sich im Forschungsstadium.
In atrophischen Bereichen verschwinden die Choriokapillaris, das RPE und die Photorezeptoren (Stäbchen und Zapfen). Die Zellzahl in der äußeren Körnerschicht (ONL) nimmt deutlich ab, und die äußere Grenzmembran (OLM) kommt in direkten Kontakt mit der Bruch-Membran. Mittelgroße bis große Aderhautgefäße bleiben auch in fortgeschrittenen Krankheitsstadien lange erhalten.
Peripherin-2 ist am Rand (Randbereich) der Scheiben der Photorezeptor-Außensegmente lokalisiert und spielt eine wesentliche Rolle bei der Scheibenbildung, Stabilisierung und Aufrechterhaltung der Randkrümmung.
Eine PRPH2-Haploinsuffizienz (Verlust einer funktionellen Kopie) beeinträchtigt die normale Bildung der Außensegmentmorphologie. Der strukturelle Zusammenbruch des Außensegments führt zur Apoptose der Photorezeptoren, was eine sekundäre Atrophie des RPE und der Choriokapillaris zur Folge hat.
Es wird angenommen, dass PRPH2 unterschiedliche Funktionen in Stäbchen und Zapfen hat. In einem PRPH2-Knockout-Mausmodell wurde berichtet, dass blaue Zapfen langsamer degenerieren als andere Zapfen. Es wird angenommen, dass dieser Unterschied zwischen Stäbchen und Zapfen zur Vielfalt der klinischen Phänotypen der Krankheit beiträgt.
Das multifokale Elektroretinogramm (mfERG) zeigt eine Funktionsminderung auch im parafovealen Bereich jenseits der atrophischen Zone, die in der Fundusuntersuchung sichtbar ist. Dies deutet darauf hin, dass die Läsionen der CACD sich weiter erstrecken als die klinisch sichtbare Atrophie.
Die Forschung zur Genersatztherapie und Gen-Editierung bei erblichen Netzhauterkrankungen, einschließlich PRPH2-Mutationen, wird weltweit vorangetrieben. Bei einer verwandten Erkrankung (Choroiderämie) wurde 2014 im Vereinigten Königreich die erste klinische Gentherapie-Studie durchgeführt.
Die Pathogenitätsanalyse jeder Mutation wird mit dem Fortschritt der Genommedizin immer präziser. Die Verbreitung der Exomsequenzierung (WES) hat die Diagnosegenauigkeit erheblich verbessert 1) und ermöglicht die Identifizierung von Mutationen bei mehr Patienten.
Es wird zunehmend auf molekularer Ebene verstanden, dass PRPH2 in Stäbchen und Zapfen unterschiedliche funktionelle Rollen spielt. Diese Erkenntnis ist wichtig, um die phänotypische Vielfalt (Stäbchen-dominante vs. Zapfen-dominante Degeneration) zu erklären und könnte die Auswahl zukünftiger Therapieziele beeinflussen.