Estágio 1
Alterações do EPR parafoveal: Alterações pigmentares puntiformes ou em placas no epitélio pigmentar da retina (EPR).
Visão e sintomas subjetivos: Frequentemente normais.

A Distrofia Coroidal Central Areolar (CACD) é uma distrofia macular hereditária que causa atrofia retinocoroidal bem delimitada na região macular. A incidência é baixa (cerca de 1 em 100.000 pessoas) e é classificada como doença rara.
O início é mais comum entre os 20 e 50 anos. O padrão de herança é principalmente autossômico dominante (AD), mas casos autossômicos recessivos (AR) e esporádicos também são relatados.
O gene causador mais frequente é o PRPH2 (cromossomo 17p13), que codifica a proteína periferina-2. A periferina-2 é essencial para a formação e estabilização dos discos do segmento externo dos fotorreceptores. A haploinsuficiência (perda de função de um alelo) é considerada o principal mecanismo.
A CACD apresenta semelhança fenotípica com doenças causadas por mutações no gene ABCA4 (como a doença de Stargardt) 1), sendo o teste genético importante para o diagnóstico diferencial. Não há relatos de complicações sistêmicas.
A DMRI geralmente surge após os 60 anos e frequentemente apresenta drusas e alterações exsudativas. A CACD tem início mais jovem até a meia-idade, com atrofia bem delimitada sem drusas. Se a mutação do PRPH2 for confirmada no teste genético, o diagnóstico de CACD pode ser estabelecido.
O principal sintoma é o escotoma central bilateral (dificuldade para ver a parte central). A redução da acuidade visual pode ocorrer precocemente, mas em alguns casos a visão permanece relativamente boa até estágios avançados.
Lesões maculares simétricas bilaterais, classificadas em 4 estágios. O nervo óptico, vasos retinianos e retina periférica são preservados. O tamanho da área atrófica é aproximadamente 2-4 vezes o diâmetro do disco óptico (DD).
Estágio 1
Alterações do EPR parafoveal: Alterações pigmentares puntiformes ou em placas no epitélio pigmentar da retina (EPR).
Visão e sintomas subjetivos: Frequentemente normais.
Estágio 2
Atrofia hipopigmentada de bordos mal definidos: Área de atrofia pálida aparece fora da fóvea.
Expansão das alterações do EPR: Atrofia com bordos ainda não nítidos é observada.
Estágio 3
Atrofia do EPR com bordos bem definidos: Área de atrofia com bordos nítidos se forma fora da fóvea.
Preservação da fóvea: Neste estágio, a fóvea permanece, e a visão é relativamente preservada.
Estágio 4
Atrofia completa envolvendo a fóvea: A área de atrofia se expande para incluir a fóvea e toda a mácula.
Deficiência visual grave: A perda extensa dos capilares coroidais, fotorreceptores e EPR causa uma diminuição acentuada da visão.
A velocidade de progressão varia muito entre os indivíduos. Geralmente, o curso é lento, e alguns casos levam décadas para atingir o estágio 4. No entanto, a taxa de progressão pode diferir conforme o genótipo, e a avaliação oftalmológica periódica é essencial. O mfERG, detalhado na seção «Diagnóstico e Métodos de Exame», é um indicador de progressão precoce.
A principal causa da CACD é a mutação no gene PRPH2. O PRPH2 codifica a periferina-2, que funciona na formação e estabilização dos discos do segmento externo dos fotorreceptores. As mutações impedem a manutenção da estrutura normal do segmento externo dos fotorreceptores.
Por ser uma herança autossômica dominante (AD), a probabilidade de transmissão do gene mutante de um dos pais para o filho é de 50%. Se houver histórico familiar, o aconselhamento genético é recomendado.
Fatores de risco adquiridos ou fatores de estilo de vida que aceleram o início ou a progressão da doença ainda não foram claramente identificados.
A CACD é predominantemente de herança autossômica dominante, e o risco de transmissão para os filhos do paciente chega a 50%. A mutação pode ser identificada por teste genético, permitindo o diagnóstico definitivo e o rastreamento familiar. Além disso, como pode ser alvo de terapia gênica no futuro, o registro da mutação é importante.
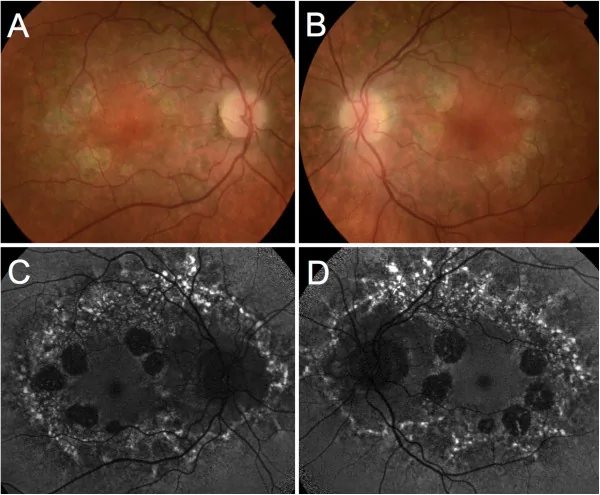
Com a lâmpada de fenda e oftalmoscópio, confirma-se uma área de atrofia bem delimitada na mácula. Avalia-se a exposição de vasos coroidais ou pigmentação.
Os achados de cada exame variam conforme o estágio da doença. A tabela abaixo mostra os achados característicos por estágio.
| Exame | Achados Iniciais | Achados Avançados |
|---|---|---|
| FAF (Autofluorescência do Fundo) | Hiperfluorescência | Perda de fluorescência (área de atrofia) |
| FA (Angiografia Fluoresceínica) | Hiperfluorescência parafoveal | Preenchimento por transparência na área de atrofia |
| OCT | Espessamento POS-RPE | Perda da camada externa (fotorreceptores e EPR) |
É importante diferenciar de doenças que apresentam atrofia macular semelhante.
| Doença Diferencial | Pontos de Diferenciação |
|---|---|
| DMRI Atrófica | Drusas, bordas irregulares, início em idade avançada |
| Doença de Stargardt | Coroide escura, mutação ABCA4 |
| Distrofia de Cones | Redução acentuada da resposta de cones no ERG, fotofobia como sintoma principal |
Atualmente, não existe tratamento estabelecido para CACD. O tratamento visa o controle dos sintomas e a manutenção da qualidade de vida.
Reabilitação para Baixa Visão
Lupas e óculos de proteção contra luz: Prescrição de auxílios para maximizar o uso da função visual residual.
Treinamento ocupacional e de vida: Apoio na aquisição de habilidades para a vida diária adaptadas à deficiência visual.
Aconselhamento Genético
Fornecimento de informações à família: Explicação sobre herança autossômica dominante (risco de 50% de herança).
Teste genético: A identificação de mutações é importante para o diagnóstico definitivo e opções futuras de tratamento.
Terapia Gênica (Fase de Pesquisa)
Candidato a tratamento futuro: Pesquisas estão em andamento sobre terapia de reposição gênica direcionada a mutações no PRPH2.
Situação atual: Ainda não pode ser oferecido como tratamento geral.
A pesquisa em terapia genética para doenças hereditárias da retina envolvendo PRPH2 está se acelerando globalmente. No Reino Unido, um ensaio de terapia genética para distrofia coroidal (coroideremia) foi realizado em 2014, e sua aplicação em doenças com mutação no PRPH2 também está sendo estudada. No entanto, atualmente não está estabelecido como tratamento padrão e ainda está em fase de pesquisa.
Na área de atrofia, os capilares coroidais, o epitélio pigmentar da retina e os fotorreceptores (bastonetes e cones) desaparecem. O número de células na camada nuclear externa diminui drasticamente, e a membrana limitante externa entra em contato direto com a membrana de Bruch. Os vasos coroidais de médio e grande porte permanecem preservados por muito tempo, mesmo em estágios avançados.
A periferina-2 está localizada na borda dos discos do segmento externo dos fotorreceptores e desempenha um papel essencial na formação, estabilização e manutenção da curvatura da borda dos discos.
Quando ocorre haploinsuficiência do PRPH2 (perda de função de uma cópia), a formação normal da morfologia do segmento externo é prejudicada. A ruptura estrutural do segmento externo progride para apoptose dos fotorreceptores, levando à atrofia secundária do epitélio pigmentar da retina e dos capilares coroidais.
Sugere-se que o PRPH2 desempenhe funções diferentes em bastonetes e cones; em camundongos knockout para PRPH2, relata-se que os cones azuis degeneram mais lentamente do que outros cones. Acredita-se que essa diferença entre bastonetes e cones contribua para a diversidade de fenótipos clínicos da doença.
Na eletrorretinografia multifocal, a diminuição da função é detectada mesmo na área parafoveal além da área de atrofia visível no exame de fundo de olho. Isso indica que as lesões da CACD se estendem mais amplamente do que a atrofia clinicamente visível.
Pesquisas sobre terapia de reposição gênica e terapia de edição gênica para doenças hereditárias da retina envolvendo mutações no PRPH2 estão sendo realizadas globalmente. Para uma doença relacionada (coroideremia), o primeiro ensaio clínico de terapia gênica foi realizado no Reino Unido em 2014.
A análise de patogenicidade de cada mutação está sendo refinada com o avanço da medicina genômica. A disseminação do sequenciamento completo do exoma (WES) melhorou significativamente a precisão diagnóstica 1), permitindo a identificação de mutações em mais pacientes.
Está sendo elucidado que o PRPH2 tem papéis funcionais diferentes em bastonetes e cones em nível molecular. Esse conhecimento é importante para explicar a diversidade fenotípica (se a degeneração é predominante em bastonetes ou cones) e pode influenciar a seleção de alvos terapêuticos no futuro.