Etapa 1
Cambios RPE parafoveales: Aparecen cambios pigmentarios punteados o parcheados en el epitelio pigmentario de la retina (RPE).
Agudeza visual y síntomas: A menudo se mantiene casi normal.

La distrofia coroidea areolar central (Central Areolar Choroidal Dystrophy; CACD) es una distrofia macular hereditaria que causa atrofia coriorretiniana bien delimitada en la mácula. Es poco frecuente (aproximadamente 1 en 100,000 personas) y se clasifica como una enfermedad rara.
La aparición es más frecuente entre los 20 y 50 años. El patrón de herencia es principalmente autosómico dominante (AD), pero también se han reportado casos autosómicos recesivos (AR) y esporádicos.
El gen causal más frecuente es PRPH2 (cromosoma 17p13), que codifica la proteína periferina-2. La periferina-2 es esencial para la formación y estabilización de los discos del segmento externo de los fotorreceptores. Se cree que la haploinsuficiencia (pérdida de función de un alelo) es el principal mecanismo de la enfermedad.
Se ha señalado que la CACD tiene similitudes fenotípicas con enfermedades causadas por mutaciones en el gen ABCA4 (como la enfermedad de Stargardt) 1), y las pruebas genéticas son importantes para el diagnóstico diferencial. No se han reportado complicaciones sistémicas.
La degeneración macular asociada a la edad generalmente ocurre después de los 60 años y a menudo presenta drusas o cambios exudativos. La CACD suele aparecer en adultos jóvenes o de mediana edad, se caracteriza por lesiones atróficas bien delimitadas y no presenta drusas. Si se confirma una mutación en PRPH2 mediante pruebas genéticas, se puede diagnosticar CACD.
El síntoma principal es el escotoma central bilateral (dificultad para ver en el centro). La agudeza visual puede disminuir tempranamente en algunos casos, pero también puede mantenerse relativamente buena hasta que la enfermedad progresa.
Ambos ojos presentan lesiones maculares simétricas, y la enfermedad se clasifica en cuatro etapas. El nervio óptico, los vasos retinianos y la retina periférica se conservan. El tamaño del área atrófica es generalmente de 2 a 4 veces el diámetro del disco óptico (DD).
Etapa 1
Cambios RPE parafoveales: Aparecen cambios pigmentarios punteados o parcheados en el epitelio pigmentario de la retina (RPE).
Agudeza visual y síntomas: A menudo se mantiene casi normal.
Etapa 2
Atrofia hipopigmentada mal definida: Aparecen áreas atróficas pálidas fuera de la fóvea.
Expansión de los cambios RPE: Se observa atrofia con bordes aún poco claros.
Etapa 3
Atrofia RPE bien definida: Se forman áreas atróficas claramente delimitadas fuera de la fóvea.
Preservación foveal: En esta etapa, la fóvea permanece intacta y la agudeza visual se conserva relativamente.
Etapa 4
Atrofia completa que afecta la fóvea: El área de atrofia se expande para incluir toda la mácula, incluida la fóvea.
Discapacidad visual grave: La pérdida extensa de la coriocapilar, los fotorreceptores y el EPR conduce a una disminución marcada de la agudeza visual.
La velocidad de progresión varía mucho entre individuos. Generalmente sigue un curso lento, y algunos casos pueden tardar décadas en llegar a la etapa 4. Sin embargo, la tasa de progresión puede diferir según el genotipo, y la evaluación oftalmológica regular es esencial. El mfERG, descrito en detalle en la sección “Diagnóstico y métodos de examen”, sirve como indicador de progresión temprana.
La causa principal de CACD son las mutaciones en el gen PRPH2. PRPH2 codifica la periferina-2, que funciona en la formación y estabilización de los discos del segmento externo de los fotorreceptores. Las mutaciones impiden el mantenimiento de la estructura normal del segmento externo de los fotorreceptores.
Debido a que es un trastorno autosómico dominante (AD), la probabilidad de heredar el gen mutado de un padre afectado a un hijo es del 50%. Si hay antecedentes familiares de la enfermedad en parientes cercanos, se recomienda asesoramiento genético.
Actualmente no se han identificado claramente factores de riesgo adquiridos o factores de estilo de vida que promuevan la aparición o progresión.
CACD es principalmente autosómico dominante, y el riesgo genético para los hijos de un paciente es del 50%. Identificar la mutación mediante pruebas genéticas permite un diagnóstico definitivo y el cribado familiar. Además, registrar la mutación es importante porque puede convertirse en un objetivo para la terapia génica futura.
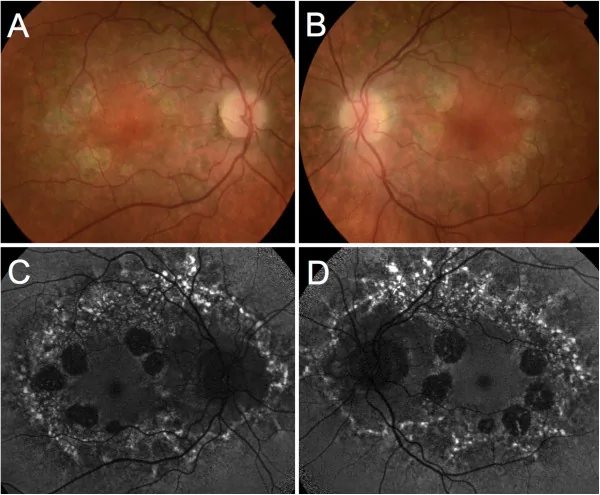
Con un microscopio de lámpara de hendidura y un oftalmoscopio, se observa un área atrófica bien delimitada en la mácula. Se evalúa la presencia de vasos coroideos expuestos y pigmentación.
Los hallazgos de cada prueba varían según la etapa de la enfermedad. La siguiente tabla muestra los hallazgos característicos por etapa.
| Prueba | Hallazgos tempranos | Hallazgos avanzados |
|---|---|---|
| FAF (autofluorescencia del fondo de ojo) | Hiperautofluorescencia | Pérdida de autofluorescencia (área atrófica) |
| FA (angiografía con fluoresceína) | Hiperfluorescencia parafoveal | Defecto de ventana en el área atrófica |
| OCT | Engrosamiento de POS-RPE | Pérdida de las capas externas (fotorreceptores y EPR) |
Es importante diferenciar de enfermedades que presentan atrofia macular similar.
| Diagnóstico diferencial | Características diferenciales clave |
|---|---|
| DMAE atrófica | Drusas, bordes irregulares, inicio tardío |
| Enfermedad de Stargardt | Coroides oscura, mutación ABCA4 |
| Distrofia de conos | Respuesta de conos en ERG marcadamente reducida, fotofobia como síntoma principal |
Actualmente, no existe un tratamiento establecido para la CACD. El tratamiento tiene como objetivo controlar los síntomas y mantener la calidad de vida.
Rehabilitación de baja visión
Lupas y gafas con filtro: Prescribir dispositivos de ayuda para maximizar la función visual restante.
Entrenamiento ocupacional y de la vida diaria: Apoyar la adquisición de habilidades para la vida diaria adaptadas a la discapacidad visual.
Consejo genético
Información para las familias: Explicar la herencia autosómica dominante (50% de riesgo genético).
Pruebas genéticas: Identificar mutaciones es importante para el diagnóstico definitivo y futuras opciones de tratamiento.
Terapia génica (en fase de investigación)
Candidato a tratamiento futuro: Se está investigando la terapia de reemplazo génico dirigida a mutaciones de PRPH2.
Estado actual: Aún no está disponible como atención médica general.
La investigación sobre terapia génica para enfermedades hereditarias de la retina, incluido PRPH2, se está acelerando a nivel mundial. En el Reino Unido, se realizó un ensayo de terapia génica para la coroideremia en 2014, y su aplicación a enfermedades con mutaciones en PRPH2 también está en estudio. Sin embargo, actualmente no está establecido como tratamiento estándar y se encuentra en fase de investigación.
En el área atrófica, desaparecen la coriocapilar, el EPR y los fotorreceptores (bastones y conos). El número de células en la capa nuclear externa (ONE) se reduce notablemente, y la membrana limitante externa (MLE) entra en contacto directo con la membrana de Bruch. Los vasos coroideos medianos y grandes se conservan durante mucho tiempo incluso en etapas avanzadas.
La periferina-2 se localiza en el borde (región del rim) de los discos del segmento externo de los fotorreceptores y desempeña un papel esencial en la formación, estabilización y mantenimiento de la curvatura del borde de los discos.
La haploinsuficiencia de PRPH2 (pérdida de una copia funcional) altera la formación normal de la morfología del segmento externo. La ruptura de la estructura del segmento externo conduce a la apoptosis de los fotorreceptores, seguida de atrofia secundaria del EPR y la coriocapilar.
Se sugiere que PRPH2 tiene funciones diferentes en bastones y conos. En modelos de ratón knockout para PRPH2, se ha informado que los conos azules degeneran más lentamente que otros conos. Se cree que esta diferencia entre bastones y conos contribuye a la diversidad de fenotipos clínicos de la enfermedad.
El electrorretinograma multifocal (mfERG) detecta una disminución funcional en el área parafoveal más allá de la zona atrófica visible en el examen de fondo de ojo. Esto indica que las lesiones de CACD se extienden más ampliamente que la atrofia clínicamente visible.
Se están llevando a cabo investigaciones a nivel mundial sobre la terapia de reemplazo génico y la terapia de edición génica para enfermedades hereditarias de la retina, incluidas las mutaciones de PRPH2. Para una enfermedad relacionada (coroideremia), el primer ensayo clínico de terapia génica se realizó en el Reino Unido en 2014.
El análisis de patogenicidad de cada mutación se está volviendo más preciso con el avance de la medicina genómica. La difusión de la secuenciación del exoma completo (WES) ha mejorado significativamente la precisión diagnóstica 1), permitiendo la identificación de mutaciones en más pacientes.
Se está dilucidando a nivel molecular que PRPH2 tiene diferentes roles funcionales en bastones y conos. Este conocimiento es importante para explicar la diversidad de fenotipos (degeneración predominante de bastones o de conos) y puede influir en la selección de futuros objetivos terapéuticos.