مرحله ۱
تغییرات RPE پارافووئال: تغییرات رنگدانهای نقطهای و لکهای در اپیتلیوم رنگدانه شبکیه (RPE) ظاهر میشود.
بینایی و علائم ذهنی: اغلب تقریباً طبیعی باقی میماند.

دیستروفی کوروئیدال مرکزی آرئولار (CACD) یک دیستروفی ماکولای ارثی است که با آتروفی مشیمیه-شبکیه با مرز مشخص در ناحیه ماکولا مشخص میشود. شیوع آن پایین است (حدود ۱ در ۱۰۰٬۰۰۰ نفر) و به عنوان یک بیماری نادر طبقهبندی میشود.
سن شروع معمولاً بین ۲۰ تا ۵۰ سالگی است. الگوی توارث عمدتاً اتوزومال غالب (AD) است، اما موارد اتوزومال مغلوب (AR) و تک گیر نیز گزارش شده است.
شایعترین ژن عامل PRPH2 (کروموزوم 17p13) است که پروتئین پریفرین-2 را کد میکند. پریفرین-2 برای تشکیل و پایداری دیسکهای بخش خارجی سلولهای بینایی ضروری است. نارسایی هاپلو (از دست دادن عملکرد یک آلل) مکتب اصلی بیماریزایی در نظر گرفته میشود.
CACD شباهت فنوتیپی با بیماریهای ناشی از جهشهای ژن ABCA4 (مانند بیماری اشتارگاردت) دارد 1) و آزمایش ژنتیک برای افتراق اهمیت دارد. عوارض سیستمیک گزارش نشده است.
دژنراسیون ماکولای وابسته به سن معمولاً بعد از ۶۰ سالگی شروع میشود و اغلب با دروزن یا تغییرات اگزوداتیو همراه است. CACD در سنین جوانی تا میانسالی شروع میشود و با ضایعات آتروفیک با مرز مشخص مشخص میشود و دروزن ندارد. در صورت تأیید جهش PRPH2 با آزمایش ژنتیک، CACD تشخیص داده میشود.
علامت اصلی اسکوتوم مرکزی دوطرفه (مشکل در دیدن مرکز) است. کاهش بینایی ممکن است زودرس باشد، اما گاهی تا مراحل پیشرفته نسبتاً خوب باقی میماند.
ضایعات ماکولار متقارن در هر دو چشم دیده میشود و بیماری به چهار مرحله تقسیم میشود. عصب بینایی، عروق شبکیه و شبکیه محیطی حفظ میشوند. اندازه ناحیه آتروفی معمولاً حدود ۲ تا ۴ برابر قطر دیسک بینایی (DD) است.
مرحله ۱
تغییرات RPE پارافووئال: تغییرات رنگدانهای نقطهای و لکهای در اپیتلیوم رنگدانه شبکیه (RPE) ظاهر میشود.
بینایی و علائم ذهنی: اغلب تقریباً طبیعی باقی میماند.
مرحله ۲
آتروفی هیپوپیگمانته با مرز نامشخص: ناحیه آتروفی کمرنگ در خارج از فووئا ظاهر میشود.
گسترش تغییرات RPE: آتروفی با مرزهای هنوز نامشخص مشاهده میشود.
مرحله ۳
آتروفی RPE با مرز مشخص: ناحیه آتروفی با مرزهای واضح در خارج از فووئا تشکیل میشود.
حفظ فووئا: در این مرحله، فووئا باقی میماند و بینایی نسبتاً حفظ میشود.
مرحله ۴
آتروفی کامل شامل حفره مرکزی: ناحیه آتروفی به کل ماکولا شامل حفره مرکزی گسترش مییابد.
اختلال شدید بینایی: به دلیل از بین رفتن گسترده لایه کوریوکاپیلاری، سلولهای بینایی و اپیتلیوم رنگدانه شبکیه، بینایی به طور قابل توجهی کاهش مییابد.
سرعت پیشرفت در افراد مختلف بسیار متفاوت است. به طور کلی سیر آهسته دارد و در برخی موارد ممکن است دهها سال طول بکشد تا به مرحله ۴ برسد. با این حال، سرعت پیشرفت ممکن است بسته به ژنوتیپ متفاوت باشد و ارزیابی منظم چشم پزشکی ضروری است. جزئیات در بخش «روشهای تشخیص و آزمایش» که mfERG به عنوان شاخص پیشرفت زودهنگام معرفی شده است، توضیح داده شده است.
علت اصلی CACD جهش در ژن PRPH2 است. PRPH2 پروتئین پریفرین-۲ را کد میکند که در تشکیل و تثبیت دیسکهای بخش خارجی سلولهای بینایی نقش دارد. جهشها مانع حفظ ساختار طبیعی بخش خارجی سلولهای بینایی میشوند.
از آنجا که این بیماری به صورت اتوزومال غالب (AD) به ارث میرسد، احتمال انتقال ژن جهشیافته از والد به فرزند ۵۰٪ است. در صورت وجود سابقه ابتلا در بستگان نزدیک، مراجعه به مشاوره ژنتیک توصیه میشود.
عوامل خطر اکتسابی یا عوامل مرتبط با سبک زندگی که باعث شروع یا تسریع پیشرفت بیماری شوند، در حال حاضر به طور قطعی شناسایی نشدهاند.
CACD عمدتاً به صورت اتوزومال غالب به ارث میرسد و خطر ژنتیکی برای فرزندان بیمار ۵۰٪ است. شناسایی جهش از طریق آزمایش ژنتیک امکان تشخیص قطعی و غربالگری خانواده را فراهم میکند. همچنین، ثبت جهش مهم است زیرا ممکن است در آینده هدف درمان ژنتیکی قرار گیرد.
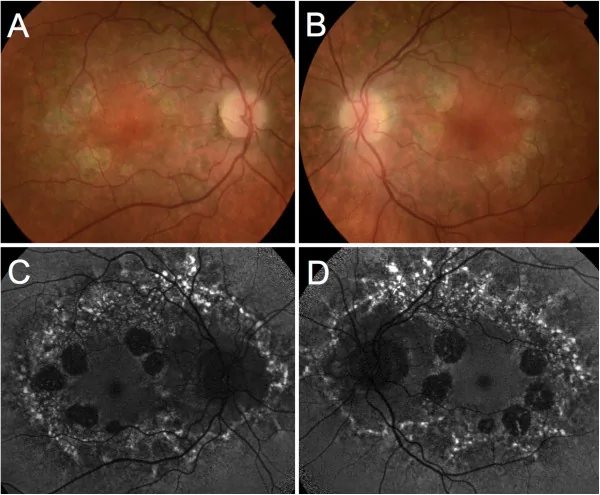
با استفاده از لامپ شکاف و افتالموسکوپ، ناحیه آتروفی با مرز مشخص در ماکولا مشاهده میشود. وجود عروق کوروئید نمایان و رسوب رنگدانه ارزیابی میگردد.
یافتههای هر آزمایش بسته به مرحله بیماری متفاوت است. جدول زیر یافتههای مشخصه هر مرحله را نشان میدهد.
| آزمایش | یافتههای اولیه | یافتههای مرحله پیشرفته |
|---|---|---|
| FAF (فلورسانس خودبخودی فوندوس) | افزایش فلورسانس | از بین رفتن فلورسانس (ناحیه آتروفی) |
| FA (آنژیوگرافی فلورسئین) | هایپرفلورسانس پارافووئال | پر شدن شفاف ناحیه آتروفی |
| OCT | ضخیم شدن POS-RPE | از بین رفتن لایه خارجی (سلولهای بینایی و RPE) |
تشخیص افتراقی از بیماریهای مشابه با آتروفی ماکوالر اهمیت دارد.
| بیماری افتراقی | نکات افتراقی |
|---|---|
| AMD آتروفیک | دروزن، مرزهای نامنظم، شروع در سنین بالا |
| بیماری اشتارگارت | مشیمیه تیره (dark choroid)، جهش ABCA4 |
| دیستروفی مخروطی | کاهش شدید پاسخ مخروطی در ERG، فتوفوبی به عنوان علامت اصلی |
در حال حاضر، درمان قطعی برای CACD وجود ندارد. هدف درمان، مدیریت علائم و حفظ کیفیت زندگی است.
توانبخشی کمبینایی
ذرهبین و عینکهای ضد نور: تجویز وسایل کمکی برای استفاده حداکثری از عملکرد بینایی باقیمانده.
آموزش شغلی و زندگی روزمره: کمک به یادگیری مهارتهای روزمره متناسب با ناتوانی بینایی.
مشاوره ژنتیک
اطلاعرسانی به خانواده: توضیح درباره وراثت اتوزومال غالب (ریسک ۵۰٪ انتقال).
آزمایش ژنتیک: شناسایی جهش برای تشخیص قطعی و انتخاب درمانهای آینده مهم است.
ژن درمانی (مرحله تحقیقاتی)
گزینه درمانی آینده: تحقیقات روی ژن درمانی جایگزین با هدف جهش PRPH2 در حال پیشرفت است.
وضعیت فعلی: در حال حاضر، این روش به عنوان درمان عمومی قابل ارائه نیست.
تحقیقات در مورد ژن درمانی برای بیماریهای ارثی شبکیه شامل PRPH2 در سراسر جهان در حال شتاب است. در بریتانیا، آزمایش ژن درمانی برای دیستروفی مشیمیه (کوروئیدرمی) در سال ۲۰۱۴ انجام شده است و کاربرد آن برای بیماریهای ناشی از جهش PRPH2 نیز در دست بررسی است. با این حال، در حال حاضر به عنوان درمان استاندارد تثبیت نشده و در مرحله تحقیقاتی قرار دارد.
در نواحی آتروفی، لایه مویرگی مشیمیه، اپیتلیوم رنگدانه شبکیه (RPE) و سلولهای گیرنده نور (میلهای و مخروطی) از بین میروند. تعداد سلولهای لایه هستهای خارجی (ONL) به شدت کاهش مییابد و غشای محدود کننده خارجی (OLM) مستقیماً با غشای بروخ تماس پیدا میکند. عروق مشیمیه با اندازه متوسط و بزرگ حتی در مراحل پیشرفته بیماری برای مدت طولانی حفظ میشوند.
پریفیرین-۲ در لبه (ناحیه rim) دیسکهای بخش خارجی سلولهای گیرنده نور قرار دارد و نقش ضروری در تشکیل، تثبیت و حفظ انحنای لبه دیسکها ایفا میکند.
هنگامی که هاپلوناکافی PRPH2 (از دست دادن یک کپی) رخ میدهد، تشکیل مورفولوژی طبیعی بخش خارجی مختل میشود. فروپاشی ساختار بخش خارجی به آپوپتوز سلولهای گیرنده نور منجر میشود و باعث آتروفی ثانویه RPE و لایه مویرگی مشیمیه میگردد.
نشان داده شده است که PRPH2 عملکردهای متفاوتی در سلولهای میلهای و مخروطی دارد و در مدل موشهای ناک اوت PRPH2، مخروطهای آبی کندتر از سایر مخروطها تحلیل میروند. تصور میشود که این تفاوت بین میلهها و مخروطها در تنوع فنوتیپ بالینی بیماری نقش دارد.
در الکترورتینوگرافی چندکانونی (mfERG)، کاهش عملکرد حتی در ناحیه پارافووه فراتر از نواحی آتروفی قابل مشاهده در معاینه فوندوس تشخیص داده میشود. این نشان میدهد که ضایعات CACD گستردهتر از آتروفی قابل مشاهده بالینی است.
تحقیقات در مورد درمان جایگزینی ژن و ویرایش ژن برای بیماریهای ارثی شبکیه شامل جهش PRPH2 در سراسر جهان در حال انجام است. در یک بیماری مرتبط (کوروئیدرمی)، اولین کارآزمایی بالینی ژن درمانی در سال ۲۰۱۴ در بریتانیا انجام شد.
تجزیه و تحلیل بیماریزایی هر جهش با پیشرفت پزشکی ژنومی دقیقتر میشود. گسترش توالییابی کامل اگزوم (WES) دقت تشخیص را به طور قابل توجهی بهبود بخشیده است 1) و امکان شناسایی جهش در بیماران بیشتری را فراهم میکند.
در حال روشن شدن است که PRPH2 نقشهای عملکردی متفاوتی در سلولهای استوانهای و مخروطی در سطح مولکولی دارد. این دانش برای توضیح تنوع فنوتیپی (تغییر غالب استوانهای در مقابل تغییر غالب مخروطی) مهم است و ممکن است بر انتخاب اهداف درمانی آینده تأثیر بگذارد.