Stadio 1
Alterazioni dell’EPR parafoveolare : compaiono alterazioni pigmentarie puntiformi o a chiazze dell’epitelio pigmentato retinico (EPR).
Acuità visiva e sintomi soggettivi : per lo più normali e stabili.

La distrofia coroideale areolare centrale (Central Areolar Choroidal Dystrophy; CACD) è una distrofia maculare ereditaria caratterizzata da atrofia corioretinica ben delimitata della macula. La frequenza è bassa (circa 1 persona su 100.000) ed è classificata tra le malattie rare.
L’esordio avviene più spesso tra i 20 e i 50 anni. La modalità di trasmissione è prevalentemente autosomica dominante (AD), ma sono stati riportati anche casi autosomici recessivi (AR) e sporadici.
Il gene causativo più frequente è PRPH2 (cromosoma 17p13), che codifica per la proteina periferina-2. La periferina-2 è essenziale per la formazione e la stabilità dei dischi dei segmenti esterni dei fotorecettori. L’aploinsufficienza (perdita di funzione di un allele) è considerata il principale meccanismo patogenetico.
La CACD mostra somiglianze fenotipiche con malattie causate da mutazioni del gene ABCA4 (come la malattia di Stargardt)1) e i test genetici sono importanti per la diagnosi differenziale. Non sono state riportate complicanze sistemiche.
L’AMD di solito insorge dopo i 60 anni e spesso si accompagna a drusen o alterazioni essudative. La CACD insorge più frequentemente in età giovane-adulta, è caratterizzata da lesioni atrofiche ben delimitate e non presenta drusen. La conferma di una mutazione PRPH2 tramite test genetico consente di diagnosticare la CACD.
Il sintomo principale è lo scotoma centrale bilaterale (difficoltà a vedere al centro). La riduzione dell’acuità visiva può essere precoce, ma talvolta una buona acuità visiva si mantiene fino a uno stadio avanzato.
Entrambi gli occhi presentano lesioni maculari simmetriche, classificate in 4 stadi. Il nervo ottico, i vasi retinici e la retina periferica sono preservati. La dimensione dell’area atrofica è generalmente di circa 2-4 diametri papillari (DD).
Stadio 1
Alterazioni dell’EPR parafoveolare : compaiono alterazioni pigmentarie puntiformi o a chiazze dell’epitelio pigmentato retinico (EPR).
Acuità visiva e sintomi soggettivi : per lo più normali e stabili.
Stadio 2
Atrofia ipopigmentata a margini sfumati : al di fuori della fovea appare un’area di atrofia pallida.
Estensione delle alterazioni dell’EPR : si osserva un’atrofia con margini ancora poco definiti.
Stadio 3
Atrofia dell’EPR a margini netti : al di fuori della fovea si forma un’area di atrofia ben delimitata.
Preservazione foveale : in questo stadio la fovea è ancora intatta e l’acuità visiva è relativamente conservata.
Stadio 4
Atrofia completa che include la fovea: l’area di atrofia si estende a tutta la macula, compresa la fovea.
Grave deficit visivo: l’estesa perdita di coriocapillari, fotorecettori ed EPR porta a una marcata riduzione dell’acuità visiva.
La velocità di progressione varia notevolmente da persona a persona. Generalmente il decorso è lento e in alcuni casi possono essere necessari decenni per raggiungere lo stadio 4. Tuttavia, la velocità di progressione può differire in base al genotipo, ed è indispensabile una valutazione oftalmologica regolare. L’mfERG, descritto in dettaglio nella sezione «Diagnosi e metodi di esame», è un indicatore di progressione precoce.
La causa principale della CACD è una mutazione nel gene PRPH2. PRPH2 codifica per la periferina-2, che funziona nella formazione e stabilizzazione dei dischi dei segmenti esterni dei fotorecettori. Le mutazioni impediscono il mantenimento della normale struttura del segmento esterno dei fotorecettori.
Poiché l’ereditarietà è autosomica dominante (AD), la probabilità di trasmissione del gene mutato da un genitore al figlio è del 50%. Se c’è una storia familiare della malattia, si raccomanda di sottoporsi a consulenza genetica.
Al momento non sono stati chiaramente identificati fattori di rischio acquisiti o legati allo stile di vita che favoriscano l’insorgenza o la progressione della malattia.
La CACD è principalmente autosomica dominante e il rischio genetico per i figli del paziente è del 50%. L’identificazione della mutazione tramite test genetico consente una diagnosi definitiva e lo screening familiare. Inoltre, poiché in futuro potrebbe essere possibile una terapia genica, è importante documentare la mutazione.
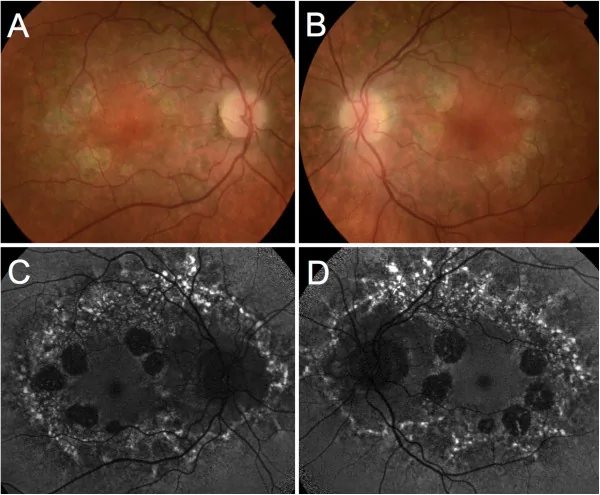
Con lampada a fessura e oftalmoscopio si conferma un’area di atrofia ben delimitata nella macula. Si valuta l’esposizione dei vasi coroideali e la presenza di depositi di pigmento.
I reperti di ciascun esame variano a seconda dello stadio della malattia. La tabella seguente mostra i reperti caratteristici per stadio.
| Esame | Reperti precoci | Reperti in fase avanzata |
|---|---|---|
| FAF (autofluorescenza del fondo) | Iperfluorescenza | Scomparsa della fluorescenza (area atrofica) |
| FA (angiografia con fluoresceina) | Iperfluorescenza parafoveale | Riempimento per trasparenza dell’area atrofica |
| OCT | Ispessimento POS-RPE | Scomparsa degli strati esterni (fotorecettori, RPE) |
È importante differenziare le malattie che presentano atrofia maculare simile.
| Malattia differenziale | Punti chiave per la differenziazione |
|---|---|
| AMD atrofica | Drusen, bordi irregolari, esordio tardivo |
| Malattia di Stargardt | Corioide scura, mutazione ABCA4 |
| Distrofia dei coni | Riduzione marcata della risposta dei coni all’ERG, fotofobia come sintomo principale |
Attualmente non esiste una terapia consolidata per la CACD. Il trattamento mira a gestire i sintomi e mantenere la qualità della vita.
Riabilitazione per ipovisione
Lenti d’ingrandimento e occhiali antiriflesso : Prescrizione di ausili per massimizzare l’uso della funzione visiva residua.
Formazione professionale e per la vita quotidiana : Supporto nell’apprendimento di tecniche di vita quotidiana adattate alla disabilità visiva.
Consulenza genetica
Informazione alle famiglie : Spiegazione della trasmissione autosomica dominante (rischio di ereditarietà del 50%).
Test genetici : L’identificazione delle mutazioni è importante per la diagnosi definitiva e le future opzioni terapeutiche.
Terapia genica (fase di ricerca)
Futuro candidato terapeutico : La ricerca sulla terapia genica sostitutiva mirata alle mutazioni PRPH2 è in corso.
Stato attuale : Al momento non è ancora disponibile come trattamento medico standard.
La ricerca sulla terapia genica per le malattie retiniche ereditarie, comprese quelle che coinvolgono PRPH2, sta accelerando a livello mondiale. Nel Regno Unito, nel 2014 è stato condotto uno studio di terapia genica per la coroideremia, e l’applicazione alle malattie da mutazione di PRPH2 è anch’essa oggetto di ricerca. Tuttavia, al momento non è ancora stabilita come trattamento standard ed è in fase di ricerca.
Nelle aree atrofiche, la coriocapillare, l’EPR e i fotorecettori (bastoncelli e coni) scompaiono. Il numero di cellule nello strato nucleare esterno (ONL) diminuisce notevolmente e la membrana limitante esterna (OLM) entra in contatto diretto con la membrana di Bruch. I vasi coroidali di medio e grande calibro vengono preservati a lungo anche negli stadi avanzati della malattia.
La periferina-2 è localizzata al bordo (regione del rim) dei dischi dei segmenti esterni dei fotorecettori e svolge un ruolo essenziale nella formazione, stabilizzazione e mantenimento della curvatura del bordo dei dischi.
L’aploinsufficienza di PRPH2 (perdita di una copia funzionale) compromette la normale formazione della morfologia del segmento esterno. La rottura strutturale del segmento esterno porta all’apoptosi dei fotorecettori, causando atrofia secondaria dell’EPR e della coriocapillare.
Si suggerisce che PRPH2 abbia funzioni diverse nei bastoncelli e nei coni. In un modello murino knockout per PRPH2, è stato riportato che i coni blu degenerano più lentamente rispetto agli altri coni. Si ritiene che questa differenza tra bastoncelli e coni contribuisca alla diversità dei fenotipi clinici della malattia.
L’elettroretinografia multifocale (mfERG) rileva una riduzione della funzione anche nell’area parafoveale oltre la zona atrofica visibile all’esame del fondo oculare. Ciò indica che le lesioni della CACD si estendono più ampiamente dell’atrofia clinicamente visibile.
La ricerca sulla terapia genica sostitutiva e sull’editing genetico per le malattie retiniche ereditarie, incluse le mutazioni PRPH2, è in corso a livello mondiale. Per una malattia correlata (coroideremia), il primo trial clinico di terapia genica è stato condotto nel Regno Unito nel 2014.
L’analisi della patogenicità di ciascuna mutazione sta diventando più precisa con il progresso della medicina genomica. La diffusione del sequenziamento dell’intero esoma (WES) ha migliorato notevolmente l’accuratezza diagnostica 1), consentendo l’identificazione di mutazioni in un numero maggiore di pazienti.
Si sta sempre più comprendendo a livello molecolare che PRPH2 ha ruoli funzionali diversi nei bastoncelli e nei coni. Questa conoscenza è importante per spiegare la diversità fenotipica (degenerazione a predominanza di bastoncelli o di coni) e potrebbe influenzare la scelta dei futuri bersagli terapeutici.