第1期
中心凹旁RPE改變:視網膜色素上皮(RPE)出現點狀或斑狀色素變化。
視力及自覺症狀:多保持正常。

中心性輪紋狀脈絡膜失養症(Central Areolar Choroidal Dystrophy; CACD)是一種遺傳性黃斑失養症,表現為黃斑部邊界清晰的視網膜脈絡膜萎縮。發生率低(約10萬人中1人),屬於罕見疾病。
發病多在20~50歲。遺傳方式以體染色體顯性遺傳(AD)為主,也有體染色體隱性遺傳(AR)和散發病例的報告。
最常見的致病基因是PRPH2(染色體17p13),編碼peripherin-2蛋白。Peripherin-2對感光細胞外節盤膜的形成和穩定至關重要。單倍劑量不足(一個等位基因功能喪失)被認為是主要發病機制。
CACD與ABCA4基因突變引起的疾病(如Stargardt病)在表型上有相似之處1),基因檢測對鑑別診斷很重要。未見全身併發症的報告。
年齡相關性黃斑部退化通常在60歲以後發病,常伴有玻璃膜疣或滲出性改變。CACD多在青中年發病,以邊界清晰的萎縮性病變為特徵,不伴有玻璃膜疣。基因檢測確認PRPH2突變即可診斷為CACD。
主要症狀是雙眼中心暗點(中心部視物不清)。視力下降有時早期即可出現,但也可能直到病程進展仍保持較好視力。
雙眼對稱性黃斑病變,病期分為4期。視神經、視網膜血管及周邊視網膜保持正常。萎縮區域大小約為視盤直徑(DD)的2~4倍。
第1期
中心凹旁RPE改變:視網膜色素上皮(RPE)出現點狀或斑狀色素變化。
視力及自覺症狀:多保持正常。
第2期
邊界不清的低色素萎縮:中心凹外出現淡色萎縮區域。
RPE變化擴大:可見邊界不清的萎縮。
第3期
邊界清晰的RPE萎縮:中心凹外形成邊界清晰的萎縮區域。
中心凹保留:此階段中心凹(fovea)尚存,視力相對保持。
第4期
累及中心凹的完全萎縮:萎縮區域擴展至包括中心凹在內的整個黃斑。
嚴重視力障礙:脈絡膜毛細血管、感光細胞和RPE廣泛喪失,導致視力顯著下降。
進展速度個體差異很大。通常病程緩慢,有些病例可能需要數十年才進入第4期。但基因型不同進展速度可能不同,定期眼科評估至關重要。詳細內容見「診斷與檢查方法」一節中描述的mfERG,可作為早期進展的指標。
CACD的主要原因是PRPH2基因突變。PRPH2編碼peripherin-2,參與感光細胞外節盤膜的形成和穩定。突變均妨礙感光細胞正常外節結構的維持。
由於是體染色體顯性遺傳(AD),攜帶突變基因的父母遺傳給子女的機率為50%。如有近親發病史,建議進行遺傳諮詢。
目前尚未明確確定後天風險因素或生活習慣可促進發病或進展。
CACD主要為體染色體顯性遺傳,患者子女的遺傳風險高達50%。透過基因檢測識別突變可實現確診和家族篩查。此外,記錄突變很重要,因為將來可能成為基因治療的對象。
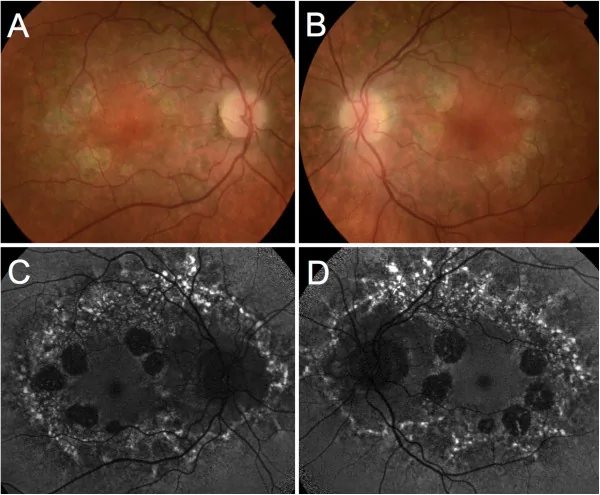
使用裂隙燈顯微鏡和眼底鏡觀察黃斑部邊界清晰的萎縮區域。評估脈絡膜血管暴露和色素沉著的存在。
根據病期不同,各檢查所見也不同。下表顯示各病期的特徵性所見。
| 檢查 | 早期所見 | 進展期所見 |
|---|---|---|
| FAF(眼底自體螢光) | 螢光增強 | 螢光消失(萎縮區域) |
| FA(螢光血管攝影) | 旁中心凹高螢光 | 萎縮區域的窗型缺損 |
| OCT | POS-RPE增厚 | 外層(感光細胞和RPE)消失 |
與呈現類似黃斑萎縮的疾病進行鑑別非常重要。
| 鑑別疾病 | 鑑別要點 |
|---|---|
| 萎縮型AMD | 玻璃膜疣、邊界不規則、老年發病 |
| Stargardt病 | 暗脈絡膜、ABCA4突變 |
| 錐體營養不良 | 視網膜電圖錐體反應顯著降低、畏光為主要症狀 |
目前,CACD尚無確立的治療方法。治療目標是控制症狀和維持生活品質。
低視力復健
放大鏡和遮光眼鏡:開立輔助器具,以最大限度地利用剩餘視功能。
職業和生活訓練:支持學習適應視覺障礙的日常生活技能。
遺傳諮詢
向家屬提供資訊:解釋體染色體顯性遺傳(50%的遺傳風險)。
基因檢測:識別突變對於確診和未來治療選擇很重要。
基因治療(研究階段)
未來的治療候選:針對PRPH2突變的基因替代療法研究正在進行中。
現狀:目前尚不能作為常規醫療提供。
在萎縮區域,脈絡膜微血管、RPE和感光細胞(視桿細胞和視錐細胞)消失。外核層(ONL)細胞數量顯著減少,外界膜(OLM)直接與布魯赫膜接觸。中大型脈絡膜血管即使在疾病晚期也能長期保留。
Peripherin-2定位於感光細胞外節盤邊緣(rim區域),在盤的形成、穩定和維持邊緣彎曲中起關鍵作用。
PRPH2單倍體不足(一個功能拷貝缺失)會損害正常外節形態的形成。外節結構破壞導致感光細胞凋亡,進而引起RPE和脈絡膜微血管的繼發性萎縮。
PRPH2在視桿細胞和視錐細胞中可能具有不同功能。在PRPH2基因剔除小鼠模型中,藍色視錐細胞的變性速度比其他視錐細胞更慢。這種視桿與視錐之間的差異被認為與疾病臨床表現型的多樣性有關。
多焦視網膜電圖(mfERG)可在眼底檢查可見的萎縮區域之外的旁中心凹區域檢測到功能下降。這表明CACD病變範圍比臨床可見的萎縮更廣泛。
針對包括PRPH2突變在內的遺傳性視網膜疾病的基因補充療法與基因編輯療法研究正在全球進行。對於相關疾病(無脈絡膜症),2014年在英國進行了首次基因治療臨床試驗。
隨著基因組醫學的進展,每種突變的致病性分析正變得更加精確。全外顯子組定序(WES)的普及大幅提高了診斷準確性1),使更多患者的突變得以識別。
PRPH2在視桿細胞和視錐細胞中具有不同功能作用的分子機制正被闡明。這項知識對於解釋表現型的多樣性(視桿細胞主導或視錐細胞主導的變性)很重要,並可能影響未來治療標靶的選擇。