Evre 1
Parafoveal RPE değişiklikleri: Retina pigment epitelinde (RPE) noktasal ve yamalı pigment değişiklikleri ortaya çıkar.
Görme ve sübjektif semptomlar: Çoğunlukla normale yakın seyreder.

Santral Areolar Koroidal Distrofi (CACD), makulada sınırları belirgin koroid-retina atrofisine neden olan kalıtsal bir makula distrofisidir. Görülme sıklığı düşüktür (yaklaşık 100.000’de 1) ve nadir hastalık olarak sınıflandırılır.
Başlangıç genellikle 20-50 yaşları arasındadır. Kalıtım paterni çoğunlukla otozomal dominanttır (AD), ancak otozomal resesif (AR) ve sporadik vakalar da bildirilmiştir.
En sık sorumlu gen PRPH2’dir (kromozom 17p13) ve periferin-2 proteinini kodlar. Periferin-2, fotoreseptör dış segment disklerinin oluşumu ve stabilizasyonu için gereklidir. Haplo-yetersizlik (bir alelin fonksiyon kaybı) ana patogenez mekanizması olarak kabul edilir.
CACD, ABCA4 gen mutasyonlarına bağlı hastalıklarla (Stargardt hastalığı gibi) fenotipik benzerlik gösterir 1) ve genetik test ayırıcı tanıda önemlidir. Sistemik komplikasyon bildirilmemiştir.
Yaşa bağlı makula dejenerasyonu genellikle 60 yaşından sonra başlar ve sıklıkla drusen veya eksüdatif değişiklikler eşlik eder. CACD genç-orta yaşta başlar, sınırları belirgin atrofik lezyonlarla karakterizedir ve drusen içermez. Genetik testle PRPH2 mutasyonu doğrulanırsa CACD tanısı konur.
Ana belirti iki taraflı merkezi skotomdur (merkezi görmenin zorlaşması). Görme azalması erken dönemde ortaya çıkabilir, ancak bazen ileri evrelere kadar nispeten iyi kalabilir.
Her iki gözde simetrik maküler lezyonlar görülür ve hastalık dört evreye ayrılır. Optik sinir, retina damarları ve periferik retina korunur. Atrofi alanının boyutu genellikle optik disk çapının (DD) yaklaşık 2-4 katıdır.
Evre 1
Parafoveal RPE değişiklikleri: Retina pigment epitelinde (RPE) noktasal ve yamalı pigment değişiklikleri ortaya çıkar.
Görme ve sübjektif semptomlar: Çoğunlukla normale yakın seyreder.
Evre 2
Sınırları belirsiz hipopigmente atrofi: Fovea dışında soluk bir atrofi alanı belirir.
RPE değişikliklerinin yayılması: Henüz net sınırları olmayan atrofi gözlenir.
Evre 3
Sınırları belirgin RPE atrofisi: Fovea dışında net sınırlı bir atrofi alanı oluşur.
Fovea korunması: Bu evrede fovea sağlam kalır ve görme nispeten korunur.
Evre 4
Fovea dahil tam atrofi: Atrofi alanı, foveayı içeren tüm makulayı kapsayacak şekilde genişler.
İleri derecede görme bozukluğu: Koroid kapiller tabakası, fotoreseptörler ve RPE’nin yaygın kaybı nedeniyle görme keskinliği belirgin şekilde azalır.
İlerleme hızı kişiden kişiye büyük farklılık gösterir. Genellikle yavaş bir seyir izler ve bazı vakalarda evre 4’e ulaşması onlarca yıl sürebilir. Ancak genotipe bağlı olarak ilerleme hızı farklılık gösterebilir ve düzenli oftalmolojik değerlendirme şarttır. Ayrıntılar, erken ilerleme göstergesi olarak mfERG’nin ele alındığı “Tanı ve Test Yöntemleri” bölümünde açıklanmıştır.
CACD’nin ana nedeni PRPH2 genindeki mutasyondur. PRPH2, periferin-2’yi kodlar ve fotoreseptör dış segment disklerinin oluşumu ve stabilizasyonunda işlev görür. Mutasyonlar, fotoreseptörlerin normal dış segment yapısının korunmasını engeller.
Otozomal dominant (AD) kalıtım olduğu için, mutasyonlu geni taşıyan ebeveynden çocuğa geçme olasılığı %50’dir. Yakın akrabalarda hastalık öyküsü varsa genetik danışmanlık alınması önerilir.
Edinsel risk faktörleri veya yaşam tarzına bağlı hastalık başlangıcını veya ilerlemesini hızlandıran faktörler şu anda net olarak tanımlanmamıştır.
CACD ağırlıklı olarak otozomal dominant kalıtım gösterir ve hastanın çocukları için genetik risk %50’dir. Genetik test ile mutasyonun belirlenmesi kesin tanı ve aile taramasını mümkün kılar. Ayrıca, gelecekte gen tedavisinin hedefi olabileceğinden mutasyonun kaydedilmesi önemlidir.
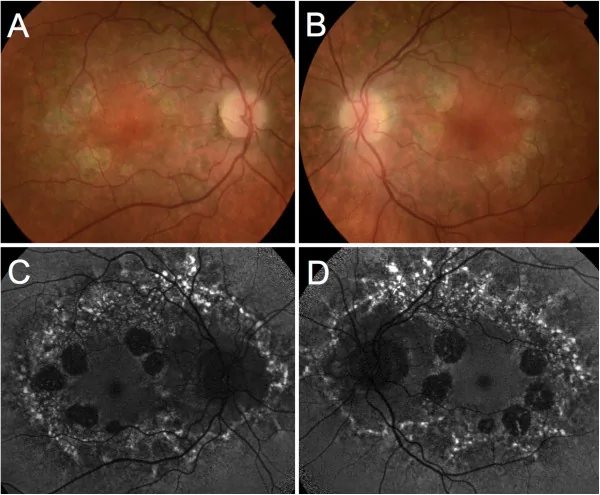
Yarık lamba biyomikroskopisi ve oftalmoskopi ile makulada sınırları belirgin atrofik alan görülür. Koroid damarlarının açığa çıkması ve pigment birikimi değerlendirilir.
Her testin bulguları hastalığın evresine göre farklılık gösterir. Aşağıdaki tablo evrelere göre karakteristik bulguları göstermektedir.
| Test | Erken dönem bulguları | İleri dönem bulguları |
|---|---|---|
| FAF (Fundus otofloresansı) | Floresans artışı | Floresans kaybı (atrofik alan) |
| FA (Floresein anjiyografi) | Parafoveal hiperfloresans | Atrofik alanda saydam dolum |
| OCT | POS-RPE kalınlaşması | Dış tabaka (fotoreseptörler ve RPE) kaybı |
Benzer maküler atrofi gösteren hastalıklarla ayırıcı tanı önemlidir.
| Ayırıcı tanı hastalığı | Ayırıcı tanı noktaları |
|---|---|
| Atrofik AMD | Drusen, düzensiz sınırlar, ileri yaş başlangıcı |
| Stargardt hastalığı | Karanlık koroid, ABCA4 mutasyonu |
| Konik distrofi | ERG konik yanıtında belirgin azalma, fotofobi ana semptom |
Şu anda CACD için kanıtlanmış bir tedavi bulunmamaktadır. Tedavi, semptomların yönetimi ve yaşam kalitesinin korunmasını hedefler.
Az Görme Rehabilitasyonu
Büyüteçler ve ışık filtreli gözlükler: Kalan görme işlevini en üst düzeyde kullanmak için yardımcı cihazlar reçete edilir.
Mesleki ve günlük yaşam eğitimi: Görme engeline uygun günlük yaşam becerilerinin kazanılması desteklenir.
Genetik Danışmanlık
Aileye bilgi verilmesi: Otozomal dominant kalıtım (%50 genetik risk) hakkında açıklama yapılır.
Genetik test: Mutasyonun belirlenmesi kesin tanı ve gelecekteki tedavi seçenekleri için önemlidir.
Gen Tedavisi (Araştırma Aşaması)
Gelecekteki tedavi adayı: PRPH2 mutasyonunu hedef alan gen yerine koyma tedavisi üzerinde araştırmalar devam etmektedir.
Mevcut durum: Şu anda genel tıbbi uygulama olarak sunulabilecek aşamada değildir.
PRPH2 dahil kalıtsal retina hastalıklarına yönelik gen tedavisi araştırmaları dünya çapında hızlanmaktadır. Birleşik Krallık’ta 2014 yılında koroidal distrofi (koroideremi) için bir gen tedavisi denemesi yapılmış olup, PRPH2 mutasyon hastalıklarına uygulanması da araştırma konusudur. Ancak şu anda standart tedavi olarak yerleşmemiştir ve araştırma aşamasındadır.
Atrofi alanlarında koroidokapillaris, RPE ve fotoreseptörler (çubuk ve koni) kaybolur. Dış nükleer tabakadaki (ONL) hücre sayısı belirgin şekilde azalır ve dış sınırlayıcı membran (OLM) doğrudan Bruch membranına temas eder. Orta ve büyük boy koroidal damarlar, hastalığın ileri evrelerinde bile uzun süre korunur.
Periferin-2, fotoreseptör dış segment disklerinin kenar (rim) bölgesinde lokalize olur ve disk oluşumu, stabilizasyonu ve kenar eğriliğinin korunmasında temel bir rol oynar.
PRPH2 haplo-yetersizliği (bir kopyanın kaybı) meydana geldiğinde, normal dış segment morfolojisinin oluşumu bozulur. Dış segment yapısının çöküşü, fotoreseptör apoptozuna ilerler ve RPE ile koroidokapillarisin sekonder atrofisine yol açar.
PRPH2’nin çubuk ve konilerde farklı işlevlere sahip olduğu öne sürülmüştür ve PRPH2 nakavt fare modelinde mavi konilerin diğer konilerden daha yavaş dejenerasyona uğradığı bildirilmiştir. Çubuk ve koniler arasındaki bu farklılığın, hastalığın klinik fenotip çeşitliliğinde rol oynadığı düşünülmektedir.
Multifokal elektroretinografide (mfERG), fundus muayenesinde görülebilen atrofi alanlarının ötesinde, parafoveal bölgede bile fonksiyon kaybı tespit edilir. Bu, CACD lezyonlarının klinik olarak görünen atrofiden daha geniş bir alana yayıldığını göstermektedir.
PRPH2 mutasyonu dahil kalıtsal retina hastalıklarına yönelik gen replasman tedavisi ve gen düzenleme tedavisi araştırmaları dünya çapında ilerlemektedir. İlgili bir hastalıkta (koroideremi), 2014 yılında Birleşik Krallık’ta ilk gen tedavisi klinik denemesi yapılmıştır.
Her mutasyonun patojenite analizi, genom tıbbındaki ilerlemelerle birlikte hassaslaşmaktadır. Tüm ekzom dizilemenin (WES) yaygınlaşması, tanı doğruluğunu önemli ölçüde artırmıştır 1) ve daha fazla hastada mutasyonların tanımlanmasını mümkün kılmaktadır.
PRPH2’nin çubuk ve koni hücrelerinde farklı fonksiyonel rollere sahip olduğu moleküler düzeyde aydınlatılmaktadır. Bu bilgi, fenotipik çeşitliliği (çubuk baskın dejenerasyon mu yoksa koni baskın dejenerasyon mu) açıklamada önemlidir ve gelecekteki tedavi hedeflerinin seçimini etkileyebilir.